www.bioin.or.kr/fileDown.do?seq=33168
/mnt/1T-5e7/mycodehtml/Biology/Gene_sequencing/Short_tutorial_text/Bioinformatics_driven_analysis_on_next_generation_DNA_sequence_data/main.html
github.com/youngminpark2559/biology/blob/master/Gene_sequencing/Next_generation_sequencing/Bioinformatics_driven_analysis_on_next_generation_DNA_sequence_data/main.html
================================================================================
/ 1970년대 생어(F. Sanger)에 의해 개발된 DNA 시퀀싱(sequencing) 기술은 인간 유전체 /
/ 프로젝트(human genome project)가 완성된 2003년경까지 독보적으로 사용되었다. 이 후 2007년경 /
/ Sanger 시퀀싱 방법을 개선한 차세대 염기서열 분석 기술(next generation sequencing, NGS) [1, 2]의 /
/ 등장으로 염기서열 해독(sequencing)에 걸리는 시간과 비용을 획기적으로 줄일 수 있게 되었다. /
/ HTS(high-throughput sequencing) 혹은 SGS(second-generation sequencing)라고도 불리는 NGS의 /
/ 가장 두드러진 특징은 초 병렬(massively parallel)적으로 백만에서 십억 단위의 리드(reads, 염기서열 /
/ 단편)를 단 몇 시간 혹은 몇 일 내로 해독할 수 있다는 것이다. /
================================================================================
1970-2003: DNA sequencing by F. Sanger
2007-: Next Generation Sequencing
Next_Generation_Sequencing
- Able to decode millions and 10 billions of reads at the same time during several hours or within several days
- which is "massively parallel" advantage
- reads: piece of DNA sequence
================================================================================
/ NGS 플랫폼(platform)을 생산하는 대표적인 회사로 Roche/454 [3], Illumina/Solexa [4], /
/ 그리고 Life/APG [5] 등을 들 수 있는데, 플랫폼은 PCR 증폭(amplification) 방식에 따라 크게 두 종류 /
/ (solid-phase amplification, emulsion PCR 등)로 나눌 수 있다. 또한 플랫폼은 다르더라도 염기서열을 /
/ 해독하는 중요한 단계인 템플릿 DNA의 준비와 증폭, 이미지 작업 및 염기서열 해독, 염기서열 /
/ 정확도 관리(quality control) 등은 공통적으로 거치는 과정이다. NGS는 비단 DNA뿐만 아니라 RNA, /
/ 특히 RNA 전사 수준(transcription level)을 측정하는데 적용할 수도 있다. /
================================================================================
Companies which produce "NGS platform"
- Roche/454, Illumina/Solexa, Life/APG
Category of company based on PCR amplification method
- solid-phase amplification
- emulsion PCR
Common procedure
- Prepare template DNA
- DNA amplification
- Image processing
- Decode DNA sequence
- Quality control on DNA sequence
Use case by NGS
- DNA level
- RNA level
- RNA transcription level
================================================================================
/ NGS 데이터는 드 노브 어셈블리(de novo assembly) [8]와 참조 어셈블리(reference assembly) /
/ 혹은 재분석(resequencing) 등 크게 두 가지 분야에 적용될 수 있다. 두 적용 분야의 가장 큰 /
/ 차이점은 참조 염기서열(reference sequence)의 유무이다. 드 노브 어셈블리는 해당 종의 유전체 /
/ 염기서열을 처음으로 해독하는 것을 말하며, 참조 어셈블리란 이미 규명된 염기서열(참조 /
/ 염기서열)에 샘플의 리드를 정렬(alignment/mapping)하는 것으로, 샘플 간의 유전변이(genetic /
/ variation) 연구에 주로 사용된다. /
================================================================================
NGS_data can be used for:
- de novo assembly
- reference assembly (or resequencing)
================================================================================
de novo assembly:
- Decode DNA sequence on the specific spicies, FOR THE FIRST TIME
- so, it has no "reference sequence"
reference assembly (or resequencing)
- it has "reference sequence"
- performs "alignment/mapping" on reads of sample
- from "found DNA sequence"
- it is much used for "genetic variation research" between samples
================================================================================
/ 재분석을 통해 추출할 수 있는 유전변이 중 가장 대표적인 것이 단일염기다형성(single /
/ nucleotide polymorphism, SNP)인데, /
================================================================================
By using resequencing
- you can extract "genetic variation"
- for example, single nucleotide polymorphism (SNP)
================================================================================
/ NGS는 보통 100개 정도의 염기로 구성된 짧은 서열 조각인 리드를 생성하여 염기서열을 /
/ 해독한다. NGS는 해독한 염기서열을 FASTQ 형식 [12]의 파일로 저장하는데, 이를 보통 원시 /
/ 데이터라 부른다. FASTQ 파일에서 SNP/Indel calling에 이르는 과정은 다양한 통계학 및 전산학적 /
/ 방법을 사용하는 여러 분석 단계를 거치는데, 이를 프로그램화한 상업용 혹은 공개 분석 /
/ 파이프라인들이 제공되고 있다 [13]. 원시 데이터에서 SNP/Indel calling에 이르는 과정은 크게 3단계, /
/ 즉 원시 데이터 생성(raw data generation), 참조 염기서열에 리드의 정렬(alignment/mapping), /
/ 그리고 유전변이 추출(variant calling) 등으로 구분할 수 있고 [14, 15], 각 단계의 결과물로 세가지 /
/ 형식(FASTQ, SAM/BAM, VCF)의 파일이 있다. /
================================================================================
In NGS:
# ================================================================================
# @ Step1
reads=create_reads(DNA)
num_bases=how_many_bases(reads)
# num_bases: around 100 bases
decoded_DNA_sequence=NGS_sequencing(num_bases)
fastq_file_as_raw_data=save_as_FASTQ_format(decoded_DNA_sequence)
# ================================================================================
# @ Step2
SAM_BAM_format_file=alignment_and_mapping(reads_of_reference_sequence)
# ================================================================================
# @ Step3
VCF_format_file=extract_genetic_variant(fastq_file_as_raw_data,SAM_BAM_format_file)
================================================================================
/또한 원시 데이터 생성과 리드의 정렬은 각 리드의 /
/ 염기서열 정보를 처리하는 단계 [그림 1의 왼쪽]에 해당하며, 유전변이 추출은 모든 리드에 대한 /
/ 정보 처리 결과를 통합하는 단계 [그림 1의 오른쪽]에 해당한다. /
================================================================================
Overall pipeline for SNP/Indel calling
 - Perform following process on "raw data", "reference sequence"
- Perform following process on "raw data", "reference sequence"
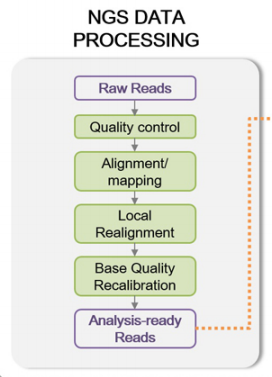 - Extract genetic variation by comparing data
- Extract genetic variation by comparing data
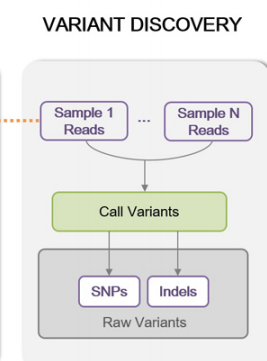 ================================================================================
/ 대부분의 파이프라인은 위의 3단계를 좀 더 세밀하게 구분(base calling and initial QC, /
/ alignment to reference, post-processing of alignment, quality score recalibration, SNP calling, filtering /
/ of SNP candidate 등)하여 데이터 분석을 진행한다 [13, 16, 17]. /
Detail steps of 3rd following "data analysis" procedure
================================================================================
/ 대부분의 파이프라인은 위의 3단계를 좀 더 세밀하게 구분(base calling and initial QC, /
/ alignment to reference, post-processing of alignment, quality score recalibration, SNP calling, filtering /
/ of SNP candidate 등)하여 데이터 분석을 진행한다 [13, 16, 17]. /
Detail steps of 3rd following "data analysis" procedure
 - base calling and initial QC
- alignment to reference
- post-processing of alignment
- quality score recalibration
- SNP calling
- filtering of SNP candidate
================================================================================
/ Broad Institute에서는 /
/ GATK(Genome Analysis Toolkit) [18]라 불리는 소위 ‘모범실무’(best practice) SNP/Indel calling /
/ 파이프라인 환경을 공개하고 있는데, GATK는 현재 NGS 데이터 분석에 가장 많이 사용되고 있는 /
/ 파이프라인이다. /
Best practice pipeline for SNP/Indel calling:
- Genome Analysis Toolkit (GATK) of Broad Institute
================================================================================
/ NGS 리드의 길이는 약 100 bp 정도로 기존 Sanger 타입의 500-1,000 bp에 비하여 길이가 /
/ 짧고, 시퀀싱 오류가 상대적으로 크며, 플랫폼에 의존하는 오류도 포함될 수 있다. /
================================================================================
NGS:
- length of read: around 100 bp
- length of read from Sanger sequencing: 500-1000 bp
- large sequencing error rate
- contains platform-driven-errors
================================================================================
/NGS 플랫폼들이 /
/ 생성하는 FASTQ 파일은 기존의 DNA 염기서열을 나타내는 텍스트 기반의 표준 염기 데이터 형식인 /
/ FASTA 형식 [19]에 해독한 염기의 정확도(quality score 혹은 error rate)를 포함시킨 것이다. /
================================================================================
FASTQ file from NGS
- FASTA data (DNA sequence)
- quality scroe (or error rate)
================================================================================
/각 리드 /
/ 당 생성되는 FASTQ 파일은 4 줄로 구성되는데, 첫째 줄은 @으로 시작하며 사용한 플랫폼과 /
/ 염기서열 길이 등에 대한 정보를 포함하고 있고, 둘째 줄은 해독한 염기서열, 셋째 줄은 + 기호로 /
/ 시작하며 기타 설명, 그리고 마지막 줄은 둘째 줄의 염기서열에 대한 정확도(quality score)를 /
/ 표시한다 [그림 2]. 따라서 둘째 줄과 넷째 줄은 같은 개수 정보로 구성된다. /
================================================================================
- base calling and initial QC
- alignment to reference
- post-processing of alignment
- quality score recalibration
- SNP calling
- filtering of SNP candidate
================================================================================
/ Broad Institute에서는 /
/ GATK(Genome Analysis Toolkit) [18]라 불리는 소위 ‘모범실무’(best practice) SNP/Indel calling /
/ 파이프라인 환경을 공개하고 있는데, GATK는 현재 NGS 데이터 분석에 가장 많이 사용되고 있는 /
/ 파이프라인이다. /
Best practice pipeline for SNP/Indel calling:
- Genome Analysis Toolkit (GATK) of Broad Institute
================================================================================
/ NGS 리드의 길이는 약 100 bp 정도로 기존 Sanger 타입의 500-1,000 bp에 비하여 길이가 /
/ 짧고, 시퀀싱 오류가 상대적으로 크며, 플랫폼에 의존하는 오류도 포함될 수 있다. /
================================================================================
NGS:
- length of read: around 100 bp
- length of read from Sanger sequencing: 500-1000 bp
- large sequencing error rate
- contains platform-driven-errors
================================================================================
/NGS 플랫폼들이 /
/ 생성하는 FASTQ 파일은 기존의 DNA 염기서열을 나타내는 텍스트 기반의 표준 염기 데이터 형식인 /
/ FASTA 형식 [19]에 해독한 염기의 정확도(quality score 혹은 error rate)를 포함시킨 것이다. /
================================================================================
FASTQ file from NGS
- FASTA data (DNA sequence)
- quality scroe (or error rate)
================================================================================
/각 리드 /
/ 당 생성되는 FASTQ 파일은 4 줄로 구성되는데, 첫째 줄은 @으로 시작하며 사용한 플랫폼과 /
/ 염기서열 길이 등에 대한 정보를 포함하고 있고, 둘째 줄은 해독한 염기서열, 셋째 줄은 + 기호로 /
/ 시작하며 기타 설명, 그리고 마지막 줄은 둘째 줄의 염기서열에 대한 정확도(quality score)를 /
/ 표시한다 [그림 2]. 따라서 둘째 줄과 넷째 줄은 같은 개수 정보로 구성된다. /
================================================================================
 - FASTQ file for "each read" is composed of 4 sentences
- FASTQ file for "each read" is composed of 4 sentences
 - First row (title identifier description) starts with @
- First row (title identifier description) starts with @
 - First row (title identifier description):
- used platform
- length of sequence
- 1: paired-end: forward (in Illumina case)
- 2: paired-end: reverse (in Illumina case)
- Second row is "decoded sequence"
- First row (title identifier description):
- used platform
- length of sequence
- 1: paired-end: forward (in Illumina case)
- 2: paired-end: reverse (in Illumina case)
- Second row is "decoded sequence"
 - Third row (as + description) starts with +
- Third row (as + description) starts with +
 - + description explains miscellaneous information
- Forth row is quality score value with respect to "decoded sequence in second row"
- + description explains miscellaneous information
- Forth row is quality score value with respect to "decoded sequence in second row"
 - Length of second row = length of forth row
================================================================================
/ 또한 Illumina 플랫폼은 /
/ 리드의 양쪽 끝에서 각각 염기서열을 해독하여 리드 당 같은 크기의 FASTQ 파일 한 쌍(forward and /
/ reverse)을 생성한다. 이것을 paired-end reads라 부른다 [그림 2]. /
================================================================================
Illumina platform:
- Decode sequence in both ends of the reads
- FASTQ_file_1_for_forward_end
- FASTQ_file_2_for_reverse_end
- paired-end reads
================================================================================
- FASTQ_file_1_for_forward_end
- Length of second row = length of forth row
================================================================================
/ 또한 Illumina 플랫폼은 /
/ 리드의 양쪽 끝에서 각각 염기서열을 해독하여 리드 당 같은 크기의 FASTQ 파일 한 쌍(forward and /
/ reverse)을 생성한다. 이것을 paired-end reads라 부른다 [그림 2]. /
================================================================================
Illumina platform:
- Decode sequence in both ends of the reads
- FASTQ_file_1_for_forward_end
- FASTQ_file_2_for_reverse_end
- paired-end reads
================================================================================
- FASTQ_file_1_for_forward_end
 - FASTQ_file_2_for_reverse_end
- FASTQ_file_2_for_reverse_end
 ================================================================================
/ FASTQ 파일이 포함하고 있는 염기서열은 SNP/Indel calling을 위한 후속 분석 과정들에 /
/ 지속적으로 영향을 미치기 때문에 염기서열의 정확도는 매우 중요하다. /
Sequence in FASTQ file
- continuously affects postprocessing for SNP/Indel calling
- accuracy of sequence in FASTQ file is important
================================================================================
/ 또한 SNP는 인간의 경우 /
/ 전체 지놈(genome)의 약 0.1% (약 1,000bp 중 1개) 정도 밖에 나타나지 않으므로 이를 확인하는 /
/ 기술은 대단히 정확해야 하며, 시퀀싱 오류는 부정확한 SNP/Indel calling으로 이어질 수 있다. /
================================================================================
SNP of human:
- 0.1% out of entire genome
- "1 bp out 1000bp" ratio
- so, detecting SNP should be highly accurate
- incorrect sequencing DNA -> incorrect detecting SNP -> incorrect SNP/Indel calling
================================================================================
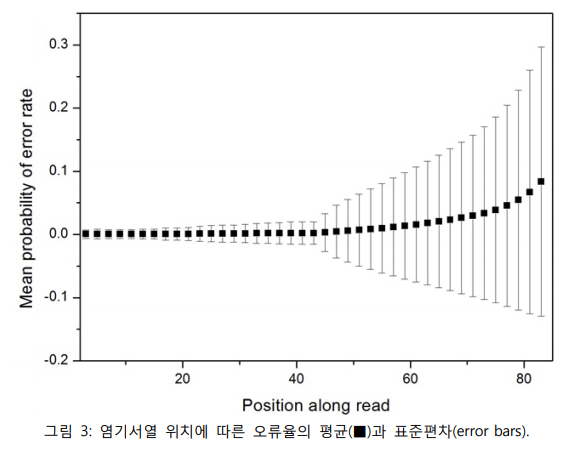
/ 일반적으로 리드 염기서열의 길이가 커짐에 따라 해독한 염기의 오류율이 증가함이 알려져 있다 [9]. /
Length of the read increases
-> decoded sequence's error rate increases
================================================================================
그림 3에서 불 수 있듯이 리드의 염기 위치(혹은 sequence cycle)가 커짐에 따라 평균 오류율은
증가하며, 특히 표준편차는 평균 오류율 보다 더 가파르게 증가한다. 이것은 sequence cycle이
증가함에 따라 오류율의 변동이 심하고, 후속 분석을 위한 데이터로 사용하기 어려운 매우 큰
오류의 염기도 포함됨을 의미한다.
================================================================================
/ FASTQ 파일이 포함하고 있는 염기서열은 SNP/Indel calling을 위한 후속 분석 과정들에 /
/ 지속적으로 영향을 미치기 때문에 염기서열의 정확도는 매우 중요하다. /
Sequence in FASTQ file
- continuously affects postprocessing for SNP/Indel calling
- accuracy of sequence in FASTQ file is important
================================================================================
/ 또한 SNP는 인간의 경우 /
/ 전체 지놈(genome)의 약 0.1% (약 1,000bp 중 1개) 정도 밖에 나타나지 않으므로 이를 확인하는 /
/ 기술은 대단히 정확해야 하며, 시퀀싱 오류는 부정확한 SNP/Indel calling으로 이어질 수 있다. /
================================================================================
SNP of human:
- 0.1% out of entire genome
- "1 bp out 1000bp" ratio
- so, detecting SNP should be highly accurate
- incorrect sequencing DNA -> incorrect detecting SNP -> incorrect SNP/Indel calling
================================================================================
/ 일반적으로 리드 염기서열의 길이가 커짐에 따라 해독한 염기의 오류율이 증가함이 알려져 있다 [9]. /
Length of the read increases
-> decoded sequence's error rate increases
================================================================================
그림 3에서 불 수 있듯이 리드의 염기 위치(혹은 sequence cycle)가 커짐에 따라 평균 오류율은
증가하며, 특히 표준편차는 평균 오류율 보다 더 가파르게 증가한다. 이것은 sequence cycle이
증가함에 따라 오류율의 변동이 심하고, 후속 분석을 위한 데이터로 사용하기 어려운 매우 큰
오류의 염기도 포함됨을 의미한다.
 ================================================================================
/ 대부분의 분석 파이프라인은 후속 분석의 정확도를 높이기 위해 NGS 염기서열의 정확도를 /
/ 관리한다. /
For high accuracy in postprocessing in sequecing pipeline,
people manage accuracy of "NGS sequence"
================================================================================
/ 염기서열의 정확도 관리는 어댑터(adaptor) 서열을 제거하거나 정확도가 현저히 낮은 염기 /
/ (예를 들어, 𝑄𝑃ℎ𝑟𝑟𝑟 < 10 인 염기)를 제거하는 것을 포함한다. /
Management of accuracy of decoded sequence
- remove adaptor
- remove sequence with low accuracy like $$$Q_{Phred} \lt 10$$$
================================================================================
/ Illumina 플랫폼의 경우에는 플랫폼 /
/ 자체적으로 부분적이나마 정확도 관리를 하고, SOLiD 플랫폼의 경우에는 추후 정렬 과정에서 /
/ 정확도가 낮은 염기는 걸러질 수 있기 때문에 특별한 정확도 관리를 하지 않는 것으로 알려져 있다 /
/ [16]. /
Illumina platform
- include accuracy-management-pipeline in their entire pipeline
SOLiD platform
- in alignment/mapping step,
- sequence which has low accuracy is removed
================================================================================
/ 현재 염기의 정확도 관리를 위한 다양한 소프트웨어가 개발되어 사용되고 있는데 [표 1], /
/ 자바(JAVA) 언어로 구현된 FastQC는 현재 가장 널리 사용되는 소프트웨어이며, qrqc은 R 언어로 /
/ 구현한 패키지이다. /
================================================================================
Software for accuracy-management
- FastQC
- most used
- Java
- qrqc
- R
================================================================================
/ 대부분의 분석 파이프라인은 후속 분석의 정확도를 높이기 위해 NGS 염기서열의 정확도를 /
/ 관리한다. /
For high accuracy in postprocessing in sequecing pipeline,
people manage accuracy of "NGS sequence"
================================================================================
/ 염기서열의 정확도 관리는 어댑터(adaptor) 서열을 제거하거나 정확도가 현저히 낮은 염기 /
/ (예를 들어, 𝑄𝑃ℎ𝑟𝑟𝑟 < 10 인 염기)를 제거하는 것을 포함한다. /
Management of accuracy of decoded sequence
- remove adaptor
- remove sequence with low accuracy like $$$Q_{Phred} \lt 10$$$
================================================================================
/ Illumina 플랫폼의 경우에는 플랫폼 /
/ 자체적으로 부분적이나마 정확도 관리를 하고, SOLiD 플랫폼의 경우에는 추후 정렬 과정에서 /
/ 정확도가 낮은 염기는 걸러질 수 있기 때문에 특별한 정확도 관리를 하지 않는 것으로 알려져 있다 /
/ [16]. /
Illumina platform
- include accuracy-management-pipeline in their entire pipeline
SOLiD platform
- in alignment/mapping step,
- sequence which has low accuracy is removed
================================================================================
/ 현재 염기의 정확도 관리를 위한 다양한 소프트웨어가 개발되어 사용되고 있는데 [표 1], /
/ 자바(JAVA) 언어로 구현된 FastQC는 현재 가장 널리 사용되는 소프트웨어이며, qrqc은 R 언어로 /
/ 구현한 패키지이다. /
================================================================================
Software for accuracy-management
- FastQC
- most used
- Java
- qrqc
- R
 ================================================================================
/ 특히 정확도가 낮은 염기를 FASTQ 파일에서 제거하는 작업을 /
/ 가지치기(trimming)라 하는데, 가지치기는 기준 염기서열에 정렬하기 전에 실시할 수도 있으며, BWA /
/ 알고리즘과 같이 정렬 과정에서 가지치기를 할 수도 있다. /
Trimming:
- remove sequences (which have low accuracy) from FASTQ file
- Scenario1
Trimming -> Alignment/mapping
- Scenario2
Trimming & Alignment/mapping at the same time
================================================================================
/ 정렬(alignment/mapping)은 유전변이와 시퀀싱 오류를 포함하고 있는 106~109 개의 /
/ 리드들을 참조 염기서열과 비교하여 리드의 염기서열과 일치하는 위치를 참조 염기서열에서 찾는 /
/ 과정이다. /
================================================================================
alignment/mapping
- you have 106-109 number of reads
- which contains "genetic variation" and "sequence error"
- you compare them with "reference sequence"
- you find the locations where "reads" and "reference sequence" are identical
================================================================================
/ 정렬에서 가장 큰 문제는 리드의 길이가 짧고 참조 염기서열 자체에 반복 영역(repetitive /
/ regions)이 있음으로 참조 염기서열의 여러 곳에 동시에 정렬될 수 있는 가능성이다 /
================================================================================
Most dangerous issue in alignment
- reads have short length
- reference sequence has long length
- which has "repetitive regions"
- therefore, "short reads" can be aligned to multiple regions of "reference sequence"
================================================================================
/ 두 염기서열을 /
/ 비교하는데 일반적으로 동적 프로그래밍(dynamic programming) 알고리즘에 기초한 BLAST(basic /
/ local alignment search tool)와 같은 소프트웨어를 사용할 수 있으나, BLAST는 계산량이 너무 많아서 /
/ NGS 데이터의 정렬에는 사용하지 않는다. /
================================================================================
To compare "reads" and "reference sequence"
- you can use "BLAST (basic local alignment search tool)"
- which is based on "dynamic programming" methodology
- but BLAST has too much computation
- so BLAST is not used to align "NGS data"
================================================================================
/ NGS 데이터의 정렬을 위한 알고리즘은 크게 /
/ BWT(Burrows-Wheeler transformation)와 해싱(hashing)에 기반한 알고리즘 등 두 가지 유형으로 /
/ 구분할 수 있는데, 두 알고리즘 모두 BLAST에 비해 계산 속도가 1,000-10,000배 정도로 빠른 /
/ 알고리즘이다. /
================================================================================
2 algorithms to align "NGS data"
- BWT (Burrows-Wheeler transformation)
- Algorithm which is based on hashing
- Both are 1000-10000x faster than BLAST
================================================================================
/ BWT는 원래 데이터 압축에 사용되는 변환으로 참조 염기서열(전체 문자열에 해당함)을 /
/ 접미사(suffix)의 특별한 순열(permutation) 형태로 변환하고, 변환된 형태에는 동일한 염기가 /
/ 연속적으로 나타나는 특징을 이용하여 리드 염기서열(부분 문자열에 해당)이 포함된 구간을 /
/ 검색한다. /
================================================================================
BWT is tranformation to compress the data
- BWT converts "reference sequence" into specific permutation form based on suffix
- Same sequence parts show repretively
- Index the parts in the "reference sequence" to which "reads" is identical
================================================================================
/ 이에 반하여 해싱 방법은 참조 염기서열과 리드 염기서열을 더 짧은 길이로 자른 후 해시 /
/ 함수(hash function)를 이용하여 해시 테이블의 값으로 표현하는 것으로, 두 염기서열의 유사성이 /
/ 해시 테이블의 같은 값으로 표현됨을 이용한 것이다. /
- Cut "reference sequence" and "reads" into more shorter lengths
- Use "hash function" to express cut "reference sequence" and "reads" as hash table value
- If 2 sequences from "reference sequence" and "reads" are similar,
- they're represented by same valu in hash table
================================================================================
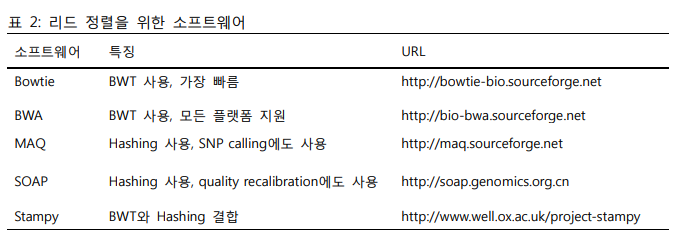
/ BWT를 사용하는 소프트웨어로 Bowtie, Bowtie2 /
/ [21], BWA [22] 등이 있으며, 해싱 방법을 사용하는 소프트웨어로 MAQ [23], Stampy [24], SOAP [25] /
/ 등이 있다 [표 2]. /
Software using BWT
- Bowtie
- Bowtie2
- BWA
Software using hasing
- MAQ
- Stampy
================================================================================
================================================================================
/ 특히 정확도가 낮은 염기를 FASTQ 파일에서 제거하는 작업을 /
/ 가지치기(trimming)라 하는데, 가지치기는 기준 염기서열에 정렬하기 전에 실시할 수도 있으며, BWA /
/ 알고리즘과 같이 정렬 과정에서 가지치기를 할 수도 있다. /
Trimming:
- remove sequences (which have low accuracy) from FASTQ file
- Scenario1
Trimming -> Alignment/mapping
- Scenario2
Trimming & Alignment/mapping at the same time
================================================================================
/ 정렬(alignment/mapping)은 유전변이와 시퀀싱 오류를 포함하고 있는 106~109 개의 /
/ 리드들을 참조 염기서열과 비교하여 리드의 염기서열과 일치하는 위치를 참조 염기서열에서 찾는 /
/ 과정이다. /
================================================================================
alignment/mapping
- you have 106-109 number of reads
- which contains "genetic variation" and "sequence error"
- you compare them with "reference sequence"
- you find the locations where "reads" and "reference sequence" are identical
================================================================================
/ 정렬에서 가장 큰 문제는 리드의 길이가 짧고 참조 염기서열 자체에 반복 영역(repetitive /
/ regions)이 있음으로 참조 염기서열의 여러 곳에 동시에 정렬될 수 있는 가능성이다 /
================================================================================
Most dangerous issue in alignment
- reads have short length
- reference sequence has long length
- which has "repetitive regions"
- therefore, "short reads" can be aligned to multiple regions of "reference sequence"
================================================================================
/ 두 염기서열을 /
/ 비교하는데 일반적으로 동적 프로그래밍(dynamic programming) 알고리즘에 기초한 BLAST(basic /
/ local alignment search tool)와 같은 소프트웨어를 사용할 수 있으나, BLAST는 계산량이 너무 많아서 /
/ NGS 데이터의 정렬에는 사용하지 않는다. /
================================================================================
To compare "reads" and "reference sequence"
- you can use "BLAST (basic local alignment search tool)"
- which is based on "dynamic programming" methodology
- but BLAST has too much computation
- so BLAST is not used to align "NGS data"
================================================================================
/ NGS 데이터의 정렬을 위한 알고리즘은 크게 /
/ BWT(Burrows-Wheeler transformation)와 해싱(hashing)에 기반한 알고리즘 등 두 가지 유형으로 /
/ 구분할 수 있는데, 두 알고리즘 모두 BLAST에 비해 계산 속도가 1,000-10,000배 정도로 빠른 /
/ 알고리즘이다. /
================================================================================
2 algorithms to align "NGS data"
- BWT (Burrows-Wheeler transformation)
- Algorithm which is based on hashing
- Both are 1000-10000x faster than BLAST
================================================================================
/ BWT는 원래 데이터 압축에 사용되는 변환으로 참조 염기서열(전체 문자열에 해당함)을 /
/ 접미사(suffix)의 특별한 순열(permutation) 형태로 변환하고, 변환된 형태에는 동일한 염기가 /
/ 연속적으로 나타나는 특징을 이용하여 리드 염기서열(부분 문자열에 해당)이 포함된 구간을 /
/ 검색한다. /
================================================================================
BWT is tranformation to compress the data
- BWT converts "reference sequence" into specific permutation form based on suffix
- Same sequence parts show repretively
- Index the parts in the "reference sequence" to which "reads" is identical
================================================================================
/ 이에 반하여 해싱 방법은 참조 염기서열과 리드 염기서열을 더 짧은 길이로 자른 후 해시 /
/ 함수(hash function)를 이용하여 해시 테이블의 값으로 표현하는 것으로, 두 염기서열의 유사성이 /
/ 해시 테이블의 같은 값으로 표현됨을 이용한 것이다. /
- Cut "reference sequence" and "reads" into more shorter lengths
- Use "hash function" to express cut "reference sequence" and "reads" as hash table value
- If 2 sequences from "reference sequence" and "reads" are similar,
- they're represented by same valu in hash table
================================================================================
/ BWT를 사용하는 소프트웨어로 Bowtie, Bowtie2 /
/ [21], BWA [22] 등이 있으며, 해싱 방법을 사용하는 소프트웨어로 MAQ [23], Stampy [24], SOAP [25] /
/ 등이 있다 [표 2]. /
Software using BWT
- Bowtie
- Bowtie2
- BWA
Software using hasing
- MAQ
- Stampy
================================================================================
 ================================================================================
/ 정렬 알고리즘의 성능은 처리량(throughput)과 정확도(accuracy)로 나타낼 수 있다. 처리량은 /
/ 정렬 속도로 같은 시간 내에 보다 많은 리드를 정렬하는 능력을 말하며, 정확도는 시퀀싱 /
/ 과정에서의 오류와 실제 염기서열의 변이에 적절히 대응하면서 리드의 정확한 정렬 위치를 /
/ 찾아내는 능력을 가리킨다. /
Performance of "alignment algorithm"
- throughput
- amount of processing reads
- accuracy
- properly responds to actual genetic variant of reads,
by comparing it to the "reference sequence"
- at the same time, it should properly respond to the error
- with distinguishing above "actual genetic variant" and "error"
- and finally, high accuracy should find the "alignment place" in reference sequence
- to which "reads" shound be aligned to
================================================================================
/ 처리량 측면에서 보면 BWT에 기반한 정렬 알고리즘이 해쉬 테이블을 /
/ 사용한 알고리즘 보다 약 10배 정도 빠르나, 정확도 측면에서는 해싱 알고리즘이 우수하다. /
/ 즉, BWT /
/ 기반 알고리즘은 변이가 적은 데이터에서는 동등한 수준의 정확도를 가지면서도 더 빠른 처리 /
/ 속도를 보이는 반면, 해싱 기반 알고리즘은 염기 변이가 많은 데이터에서 더 많은 리드를 /
/ 성공적으로 정렬한다 [24] /
- Amount of aligning "reads"
BWT is 10x faster tha hasing
- Accuracy is better in hashing than BWT
- Good case for BWT
- target data which has low number of genetic variants
- Good case for hashing
- target data which has many number of genetic variants
================================================================================
/ 특히 paired-end /
/ read의 경우에는 forward 리드와 reverse 리드 각각을 독립적으로 정렬하고 각 리드가 정렬된 /
/ 위치에 대한 정보(.sai 파일)를 구한 후, 이를 이용하여 두 리드의 정렬 결과를 합친다. /
================================================================================
paired-end read case
- align forward read, create .sai file which has information of alignment of read
- align reverse read, create .sai file which has information of alignment of read
- merge 2 alignment results
================================================================================
/ 정렬 알고리즘 선택보다 더 중요한 것은 매개변수(parameter) 값의 결정이다. /
================================================================================
Your choice
- alignment algorithm
- parameter values in selected alignment algorithm
================================================================================
/ 중요한 /
/ 매개변수로 참조 염기서열과 리드 사이의 불일치(mismatch) 염기 개수를 들 수 있다. 불일치를 /
/ 허용하지 않고 완벽한 일치만 허용한다면 후속 분석에서는 SNP/Indel은 검출할 수 없게 된다. 이와 /
/ 반대로 너무 큰 불일치를 허용하면 잘못된 정렬 결과를 나을 수 있고, 그 결과로 많은 /
/ 거짓양성(false-positive) SNP/Indel을 얻게 된다. 따라서 적절한 불일치 염기 개수를 결정하는 것이 /
/ 중요한 문제로 부각된다. /
Mismatch parameter:
- mismatching threshold value between "reference sequence" and "read"
- too low mismatching threshold value (perfect matching)
- in postprocessing, you can't find "SNP/Indel"
- too high mismatching threshold value (losen matching criterion)
- can create incorrect alignment result (many false-positive SNP/Indel result)
================================================================================
유전체는 리드의 길이와 유사하거나 더 긴 길이의 반복 염기서열(repetitive
sequence)이 존재하기 때문에, 하나의 리드가 참조 염기서열의 여러 위치에 동일한 정확도로 정렬될
수 있다. 또한 몇 개의 돌연변이(mutation) 혹은 오류로 인하여 다른 위치에 정렬될 수 있다. 따라서
정렬 과정이 끝난 다음 정렬 결과를 다양한 통계치를 사용하여 검토하는 것이 필요하다. 예를 들면
기준 염기서열에 정렬된 리드의 비율, paired-end 리드의 경우에는 성공적으로 쌍(pair)을 이룬 비율,
그리고 paired-end의 삽입 크기(insert size)에 대한 빈도 등을 들 수 있다. 이러한 통계치는 정렬을
위한 매개변수 결정에 도움을 줄 수 있으며, 정확도를 높이는데 기여할 수 있다.
================================================================================
/ 리드들의 정렬 결과는 SAM(sequence alignment map) 형식 [26]으로 저장된다. /
Alignment result of "reads" is saved into SAM (sequence alignment map) file
================================================================================
/ SAM 파일은 /
/ 최소 11개의 필드는 반드시 가지고 있는데, 리드이름, 리드가 참조 염기서열에 정렬된 결과를 /
/ 나타내는 bitwise flags, 참조 염기서열 이름, 정렬된 위치, 정렬 정확도(mapping quality), CIGAR 등의 /
/ 정보를 포함한다 [그림 4].
================================================================================
SAM file has at least 11 number of fields
SAM file contains following information
- name of read
- bitwise flags: alignment result of reads "into reference sequence"
- name of reference sequence
- locations of alignment
- accuracy of alignment (or mapping quality)
- CIGAR
================================================================================
SAM/BAM file example
================================================================================
/ 정렬 알고리즘의 성능은 처리량(throughput)과 정확도(accuracy)로 나타낼 수 있다. 처리량은 /
/ 정렬 속도로 같은 시간 내에 보다 많은 리드를 정렬하는 능력을 말하며, 정확도는 시퀀싱 /
/ 과정에서의 오류와 실제 염기서열의 변이에 적절히 대응하면서 리드의 정확한 정렬 위치를 /
/ 찾아내는 능력을 가리킨다. /
Performance of "alignment algorithm"
- throughput
- amount of processing reads
- accuracy
- properly responds to actual genetic variant of reads,
by comparing it to the "reference sequence"
- at the same time, it should properly respond to the error
- with distinguishing above "actual genetic variant" and "error"
- and finally, high accuracy should find the "alignment place" in reference sequence
- to which "reads" shound be aligned to
================================================================================
/ 처리량 측면에서 보면 BWT에 기반한 정렬 알고리즘이 해쉬 테이블을 /
/ 사용한 알고리즘 보다 약 10배 정도 빠르나, 정확도 측면에서는 해싱 알고리즘이 우수하다. /
/ 즉, BWT /
/ 기반 알고리즘은 변이가 적은 데이터에서는 동등한 수준의 정확도를 가지면서도 더 빠른 처리 /
/ 속도를 보이는 반면, 해싱 기반 알고리즘은 염기 변이가 많은 데이터에서 더 많은 리드를 /
/ 성공적으로 정렬한다 [24] /
- Amount of aligning "reads"
BWT is 10x faster tha hasing
- Accuracy is better in hashing than BWT
- Good case for BWT
- target data which has low number of genetic variants
- Good case for hashing
- target data which has many number of genetic variants
================================================================================
/ 특히 paired-end /
/ read의 경우에는 forward 리드와 reverse 리드 각각을 독립적으로 정렬하고 각 리드가 정렬된 /
/ 위치에 대한 정보(.sai 파일)를 구한 후, 이를 이용하여 두 리드의 정렬 결과를 합친다. /
================================================================================
paired-end read case
- align forward read, create .sai file which has information of alignment of read
- align reverse read, create .sai file which has information of alignment of read
- merge 2 alignment results
================================================================================
/ 정렬 알고리즘 선택보다 더 중요한 것은 매개변수(parameter) 값의 결정이다. /
================================================================================
Your choice
- alignment algorithm
- parameter values in selected alignment algorithm
================================================================================
/ 중요한 /
/ 매개변수로 참조 염기서열과 리드 사이의 불일치(mismatch) 염기 개수를 들 수 있다. 불일치를 /
/ 허용하지 않고 완벽한 일치만 허용한다면 후속 분석에서는 SNP/Indel은 검출할 수 없게 된다. 이와 /
/ 반대로 너무 큰 불일치를 허용하면 잘못된 정렬 결과를 나을 수 있고, 그 결과로 많은 /
/ 거짓양성(false-positive) SNP/Indel을 얻게 된다. 따라서 적절한 불일치 염기 개수를 결정하는 것이 /
/ 중요한 문제로 부각된다. /
Mismatch parameter:
- mismatching threshold value between "reference sequence" and "read"
- too low mismatching threshold value (perfect matching)
- in postprocessing, you can't find "SNP/Indel"
- too high mismatching threshold value (losen matching criterion)
- can create incorrect alignment result (many false-positive SNP/Indel result)
================================================================================
유전체는 리드의 길이와 유사하거나 더 긴 길이의 반복 염기서열(repetitive
sequence)이 존재하기 때문에, 하나의 리드가 참조 염기서열의 여러 위치에 동일한 정확도로 정렬될
수 있다. 또한 몇 개의 돌연변이(mutation) 혹은 오류로 인하여 다른 위치에 정렬될 수 있다. 따라서
정렬 과정이 끝난 다음 정렬 결과를 다양한 통계치를 사용하여 검토하는 것이 필요하다. 예를 들면
기준 염기서열에 정렬된 리드의 비율, paired-end 리드의 경우에는 성공적으로 쌍(pair)을 이룬 비율,
그리고 paired-end의 삽입 크기(insert size)에 대한 빈도 등을 들 수 있다. 이러한 통계치는 정렬을
위한 매개변수 결정에 도움을 줄 수 있으며, 정확도를 높이는데 기여할 수 있다.
================================================================================
/ 리드들의 정렬 결과는 SAM(sequence alignment map) 형식 [26]으로 저장된다. /
Alignment result of "reads" is saved into SAM (sequence alignment map) file
================================================================================
/ SAM 파일은 /
/ 최소 11개의 필드는 반드시 가지고 있는데, 리드이름, 리드가 참조 염기서열에 정렬된 결과를 /
/ 나타내는 bitwise flags, 참조 염기서열 이름, 정렬된 위치, 정렬 정확도(mapping quality), CIGAR 등의 /
/ 정보를 포함한다 [그림 4].
================================================================================
SAM file has at least 11 number of fields
SAM file contains following information
- name of read
- bitwise flags: alignment result of reads "into reference sequence"
- name of reference sequence
- locations of alignment
- accuracy of alignment (or mapping quality)
- CIGAR
================================================================================
SAM/BAM file example
 ================================================================================
/ 정렬 여부에 대한 중요한 정보는 bitwise flags에 포함되어 있는데, 모든 /
/ 리드가 기준 염기서열에 유일하고 정확하게 정렬되는 것은 아님으로 다양한 유형의 정렬 결과가 /
/ 가능하고 중요한 유형을 표 3에 요약하였다. /
================================================================================
Important information about the alignment
- bigwise flags
- All reads are not always aligned to "reference sequence" uniquely and precisely
- it means there can be various types of alignment
================================================================================
Most important 3 types of alignment
================================================================================
/ 정렬 여부에 대한 중요한 정보는 bitwise flags에 포함되어 있는데, 모든 /
/ 리드가 기준 염기서열에 유일하고 정확하게 정렬되는 것은 아님으로 다양한 유형의 정렬 결과가 /
/ 가능하고 중요한 유형을 표 3에 요약하였다. /
================================================================================
Important information about the alignment
- bigwise flags
- All reads are not always aligned to "reference sequence" uniquely and precisely
- it means there can be various types of alignment
================================================================================
Most important 3 types of alignment
 Type1: read are uniquely mapped to reference sequence
When: accuracy of read's sequence is high
Type2: read are ambiguously mapped to reference sequence
When: reads are mapped to "repetitive sequence" of "reference sequence"
when "reference sequence" comes from "homologuous region"
Type3: read are not mapped to reference sequence
When: sample for reads contains "chromosome" of other species
when reads contain "adapter"
when sequence in reads had been ambiguously sequenced
Error from PCR step
when reads are affected by structural variation
================================================================================
CIGAR는 정렬된 서열에 대한 정보를 부호화한 것으로,
정렬된 염기의 개수, insertion, deletion, skipped region에 관한 정보 등을 표현한다.
================================================================================
CIGAR:
- encodes information of aligned sequence
- contains information of
- number of aligned sequence (read)
- insertion
- deletion
- skipped region
================================================================================
/ 또한 SAM /
/ 파일을 이진(binary) 형식으로 압축한 BAM 파일은 SAM 파일과 더불어 현재 정렬 결과를 저장하는 /
/ 표준형식으로 자리잡고 있다. /
================================================================================
BAM_file = converter_into_binary_file(SAM_file)
SAM_file, BAM_file
- Standard file format to save "result of alignment"
================================================================================
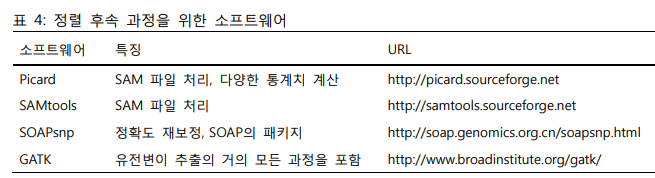
/ SAM/BAM 파일을 효율적으로 처리할 수 있는 소프트웨어로 SAMtools /
/ [26], GATK [27], Picard [28] 등이 현재 많이 사용되고 있다. BAM 파일은 IGV(Integrative Genome /
/ Viewer) 소프트웨어 [29]를 통해 시각화할 수 있으며, CRAMTools 패키지 [30]을 사용하여 CRAM /
/ 형식으로 압축할 수도 있다. /
================================================================================
Softwares to process SAM/BAM files
- SAMtools
- GATK
- Picard
================================================================================
/ BAM 파일은 IGV(Integrative Genome /
/ Viewer) 소프트웨어 [29]를 통해 시각화할 수 있으며, CRAMTools 패키지 [30]을 사용하여 CRAM /
/ 형식으로 압축할 수도 있다. /
================================================================================
Visualization on BAM file
- IGV (Integrative Genome Viewer) software
Compress BAM file into CRAM file
- CRAMTools
================================================================================
/ BAM/SAM 파일을 유전변이 추출(variant calling) 단계에 적용 하기 전에 일반적으로 두 가지 /
/ 선행 처리, 즉 정렬 후속 과정(alignment post processing)과 염기 정확도 재보정(quality score /
/ recalibration)을 수행한다. /
================================================================================
BAM/SAM
-> alignment post processing
-> quality score recalibration
-> variant calling (extracting genetic variant)
================================================================================
/ 정렬 후속 과정은 정렬된 결과를 염색체 별로 분류하고, PCR 과정에서 /
/ 발생하는 인공적 결함(artifact)를 제거하며, 참조 염기서열의 여러 위치에 동시에 정렬된 리드들을 /
/ 제거하는 것을 포함하고, Indel을 중심으로 지역적 재정렬(local realignment)을 실행하기도 한다. /
/ 정렬 후속 과정은 소프트웨어 SAMtools 혹은 Picard [표 4]등으로 처리할 수 있다. /
================================================================================
alignment post processing
- Classify "aligned sequence of read" into "chromosomes"
- Remove artifacts which are created in PCR
- Remove "reads" which are aligned to multiple locations in "reference sequence"
- Perform "local realignment" with considering "Indel"
- Software
- SAMtools
- Picard
Type1: read are uniquely mapped to reference sequence
When: accuracy of read's sequence is high
Type2: read are ambiguously mapped to reference sequence
When: reads are mapped to "repetitive sequence" of "reference sequence"
when "reference sequence" comes from "homologuous region"
Type3: read are not mapped to reference sequence
When: sample for reads contains "chromosome" of other species
when reads contain "adapter"
when sequence in reads had been ambiguously sequenced
Error from PCR step
when reads are affected by structural variation
================================================================================
CIGAR는 정렬된 서열에 대한 정보를 부호화한 것으로,
정렬된 염기의 개수, insertion, deletion, skipped region에 관한 정보 등을 표현한다.
================================================================================
CIGAR:
- encodes information of aligned sequence
- contains information of
- number of aligned sequence (read)
- insertion
- deletion
- skipped region
================================================================================
/ 또한 SAM /
/ 파일을 이진(binary) 형식으로 압축한 BAM 파일은 SAM 파일과 더불어 현재 정렬 결과를 저장하는 /
/ 표준형식으로 자리잡고 있다. /
================================================================================
BAM_file = converter_into_binary_file(SAM_file)
SAM_file, BAM_file
- Standard file format to save "result of alignment"
================================================================================
/ SAM/BAM 파일을 효율적으로 처리할 수 있는 소프트웨어로 SAMtools /
/ [26], GATK [27], Picard [28] 등이 현재 많이 사용되고 있다. BAM 파일은 IGV(Integrative Genome /
/ Viewer) 소프트웨어 [29]를 통해 시각화할 수 있으며, CRAMTools 패키지 [30]을 사용하여 CRAM /
/ 형식으로 압축할 수도 있다. /
================================================================================
Softwares to process SAM/BAM files
- SAMtools
- GATK
- Picard
================================================================================
/ BAM 파일은 IGV(Integrative Genome /
/ Viewer) 소프트웨어 [29]를 통해 시각화할 수 있으며, CRAMTools 패키지 [30]을 사용하여 CRAM /
/ 형식으로 압축할 수도 있다. /
================================================================================
Visualization on BAM file
- IGV (Integrative Genome Viewer) software
Compress BAM file into CRAM file
- CRAMTools
================================================================================
/ BAM/SAM 파일을 유전변이 추출(variant calling) 단계에 적용 하기 전에 일반적으로 두 가지 /
/ 선행 처리, 즉 정렬 후속 과정(alignment post processing)과 염기 정확도 재보정(quality score /
/ recalibration)을 수행한다. /
================================================================================
BAM/SAM
-> alignment post processing
-> quality score recalibration
-> variant calling (extracting genetic variant)
================================================================================
/ 정렬 후속 과정은 정렬된 결과를 염색체 별로 분류하고, PCR 과정에서 /
/ 발생하는 인공적 결함(artifact)를 제거하며, 참조 염기서열의 여러 위치에 동시에 정렬된 리드들을 /
/ 제거하는 것을 포함하고, Indel을 중심으로 지역적 재정렬(local realignment)을 실행하기도 한다. /
/ 정렬 후속 과정은 소프트웨어 SAMtools 혹은 Picard [표 4]등으로 처리할 수 있다. /
================================================================================
alignment post processing
- Classify "aligned sequence of read" into "chromosomes"
- Remove artifacts which are created in PCR
- Remove "reads" which are aligned to multiple locations in "reference sequence"
- Perform "local realignment" with considering "Indel"
- Software
- SAMtools
- Picard
 ================================================================================
/ SOAPsnp는 /
/ 시퀸싱 플랫폼이 제공한 염기 정확도, 리드에서 염기의 위치(sequencing cycle), 대체 /
/ 유형(substitution type, 예: 참조 염기서열에서는 A 인데 리드에서는 G인 경우) 등을 사용하여 참조 /
/ 염기서열에 대하여 평균 불일치 비율(mismatch rate)를 계산하고, 그 결과를 보정된 염기 /
/ 정확도(recalibrated quality score)를 결정하는데 사용한다. /
================================================================================
quality score recalibration by SOAPsnp software
- Use following infromation to calculate "average mismatch rate"
between "read" and "reference sequence"
- Accuracy of sequence which is provided from "sequencing platform"
- locations of base in read (or sequencing cycle)
- substitution type
- for example, case where A in reference sequence, but G in read
- Calculate result is used to determine "recalibrated quality score"
================================================================================
/ GATK도 유사한 방법을 사용하는데, 염기를 /
/ 정확도와 dinucleotide content 등으로 실험적인 오류 모델을 사용하여 분류한 후, 각 분류 군에 /
/ 대하여 실험적인 불일치 비율을 계산하여 보정된 염기 정확도를 결정하는 자료로 사용한다. /
================================================================================
quality score recalibration by GATK software
- Classify "base (or sequence)"
by using "accuracy" and "dinucleotide content"
- Calculate "mismatching ratio" with respect to each class
- Calculated ratio is used to determine accuracy of quality score recalibration
================================================================================
/ 정렬과 후속 과정을 마친 염기서열 데이터의 특성은 너비(breath)와 깊이(depth 혹은 /
/ coverage) 등 크게 두 가지 척도로 표현할 수 있다. 너비란 규명한 유전체 정도를 나타내며, 깊이란 /
/ 유전체에서 각 염기가 리드에 의해 평균적으로 규명된 정도를 나타낸다. 깊이는 염기에 대하여 /
/ 대략적으로 정규분포를 따른다. /
================================================================================
After, "alignment" and "postprocessing (alignment post processing quality score recalibration)"
sequence data can be expressed in "breath" and "depth (or coverage)"
Depth follows "Normal distribution" when "base" is variable
================================================================================
/ 변이 추출(variant calling)이란 정렬, 정렬 후속 과정, 그리고 염기 정확도 재보정 등을 거친 /
/ 리드들의 BAM/SAM 파일들을 통합하여 SNP/Indel 영역을 찾는 과정이다. /
================================================================================
Variation calling (extracting genetic variant from read)
alignment
-> postprocessing after alignment
-> quality score recalibration
-> variation calling
variation calling
- Merge BAM/SAM files of reads
- Find regions of SNP/Indel
================================================================================
/ 변이 추출은 염기서열의 /
/ 위치를 SNP/Indel로 확인될 확률로 표현하며, 주로 베이지안 방법(Bayesian method) [25]으로 /
/ 계산한다. /
================================================================================
variation calling expresses location of sequence
as probability of "that sequence" confirmed as SNP/Indel
Calculation:
- Bayesian method
================================================================================
/ 베이지안 방법이란 사전확률(prior probability)과 우도(likelihood)를 사용하여 /
/ 사후확률(posterior probability)을 계산하는 방법으로, 단순히 우도만 사용하여 추정하는 것보다 더 /
/ 많은 정보를 얻을 수 있다. /
================================================================================
Bayesian method:
- Calculate posterior probability
- by using prior probability and likelihood
- By using Bayesian method,
- you can get more information
- than the method where you use only likelihood
================================================================================
/ 이때 사전확률은 선험적으로 각 염기 위치(base position)가 유전변이가 /
/ 될 확률이며, 우도는 그 위치의 염기가 서로 다른 유전자형이 될 빈도(frequency)이다. /
================================================================================
prior probability
- probability of each base position becoming "genetic variation"
likelihood
- frequency of specific location's base becoming differening genetic type
================================================================================
/ 즉, 어느 /
/ locus에서 샘플 𝑖의 유전자형이 𝐺𝑖가 될 확률은 NGS 데이터 집합 𝐷와 베이즈 규칙(Bayes’ rule)에 /
/ 의하여 /
================================================================================
$$$p(G_i|D)
= \dfrac{p(G_i) p(D|G_i)}{ \sum\limits_{k=1}^{S} p(G_k) p(D|G_k)}$$$
$$$D$$$: NGS data set
$$$i$$$: ith sample from locus
$$$G_i$$$: genetic type of sample i
$$$S$$$: number of genetic type
$$$p(G_i)$$$: prior probability
$$$p(D|G_i)$$$: likelihood
- prior probability $$$p(G_i)$$$ is predicted
by using SNP database like dbSNP
================================================================================
/ 변이 추출 결과는 VCF(variant call format) [32] 형식의 파일에 저장된다. /
================================================================================
Result from variation calling (extracting genetic variant from read)
- is saved into VCF (variant call format) file
================================================================================
/ VCF 파일은 /
/ SNP/Indel로 확인된 유전변이 후보에 대한 정보, 즉 염색체 위치, 참조 염기, 치환/삽입/삭제 등이 /
/ 일어난 표본 염기, 변이 정확도(variation quality), 변이로 판명된 샘플의 빈도 등을 포함하고 있다. /
================================================================================
VCF file
- contains following information which is confirmed via SNP/Indel
- information about candidate genetic variation
- location of chromosome (of genetic variant occured?)
- reference sequence
- sample sequence which is occurred by
- substitution, insertion, deletion
- variation quality
- frequency of sample which is confirmed as genetic variation
================================================================================
/ 또한 변이 빈도(variation frequency)에 의해 SNP는 homozygous와 heterozygous 등 두 가지 /
/ 유형으로 나뉜다. Homozygous는 해당 위치에 정렬된 거의 모든 샘플에서 변이가 일어난 경우이고, /
/ heterozygous는 일부 샘플에서만 변이가 일어난 경우이다. 그림 5는 SAMtools를 사용하여 구한 /
/ VCF 형식의 파일이다. /
================================================================================
SNP:
- has following 2 types due to variation frequency
- homozygous
- heterozygous
================================================================================
/ Homozygous는 해당 위치에 정렬된 거의 모든 샘플에서 변이가 일어난 경우이고, /
/ heterozygous는 일부 샘플에서만 변이가 일어난 경우이다. 그림 5는 SAMtools를 사용하여 구한 /
/ VCF 형식의 파일이다.
================================================================================
Homozygous:
- Almost all samples which are aligned to reference sequence
have genetic variation
heterozygous:
- Only some part of samples which are aligned to reference sequence
have genetic variation
================================================================================
/ 그림 5는 SAMtools를 사용하여 구한 /
/ VCF 형식의 파일이다. /
================================================================================
VCF file example which is obtained via SAMtools
================================================================================
/ SOAPsnp는 /
/ 시퀸싱 플랫폼이 제공한 염기 정확도, 리드에서 염기의 위치(sequencing cycle), 대체 /
/ 유형(substitution type, 예: 참조 염기서열에서는 A 인데 리드에서는 G인 경우) 등을 사용하여 참조 /
/ 염기서열에 대하여 평균 불일치 비율(mismatch rate)를 계산하고, 그 결과를 보정된 염기 /
/ 정확도(recalibrated quality score)를 결정하는데 사용한다. /
================================================================================
quality score recalibration by SOAPsnp software
- Use following infromation to calculate "average mismatch rate"
between "read" and "reference sequence"
- Accuracy of sequence which is provided from "sequencing platform"
- locations of base in read (or sequencing cycle)
- substitution type
- for example, case where A in reference sequence, but G in read
- Calculate result is used to determine "recalibrated quality score"
================================================================================
/ GATK도 유사한 방법을 사용하는데, 염기를 /
/ 정확도와 dinucleotide content 등으로 실험적인 오류 모델을 사용하여 분류한 후, 각 분류 군에 /
/ 대하여 실험적인 불일치 비율을 계산하여 보정된 염기 정확도를 결정하는 자료로 사용한다. /
================================================================================
quality score recalibration by GATK software
- Classify "base (or sequence)"
by using "accuracy" and "dinucleotide content"
- Calculate "mismatching ratio" with respect to each class
- Calculated ratio is used to determine accuracy of quality score recalibration
================================================================================
/ 정렬과 후속 과정을 마친 염기서열 데이터의 특성은 너비(breath)와 깊이(depth 혹은 /
/ coverage) 등 크게 두 가지 척도로 표현할 수 있다. 너비란 규명한 유전체 정도를 나타내며, 깊이란 /
/ 유전체에서 각 염기가 리드에 의해 평균적으로 규명된 정도를 나타낸다. 깊이는 염기에 대하여 /
/ 대략적으로 정규분포를 따른다. /
================================================================================
After, "alignment" and "postprocessing (alignment post processing quality score recalibration)"
sequence data can be expressed in "breath" and "depth (or coverage)"
Depth follows "Normal distribution" when "base" is variable
================================================================================
/ 변이 추출(variant calling)이란 정렬, 정렬 후속 과정, 그리고 염기 정확도 재보정 등을 거친 /
/ 리드들의 BAM/SAM 파일들을 통합하여 SNP/Indel 영역을 찾는 과정이다. /
================================================================================
Variation calling (extracting genetic variant from read)
alignment
-> postprocessing after alignment
-> quality score recalibration
-> variation calling
variation calling
- Merge BAM/SAM files of reads
- Find regions of SNP/Indel
================================================================================
/ 변이 추출은 염기서열의 /
/ 위치를 SNP/Indel로 확인될 확률로 표현하며, 주로 베이지안 방법(Bayesian method) [25]으로 /
/ 계산한다. /
================================================================================
variation calling expresses location of sequence
as probability of "that sequence" confirmed as SNP/Indel
Calculation:
- Bayesian method
================================================================================
/ 베이지안 방법이란 사전확률(prior probability)과 우도(likelihood)를 사용하여 /
/ 사후확률(posterior probability)을 계산하는 방법으로, 단순히 우도만 사용하여 추정하는 것보다 더 /
/ 많은 정보를 얻을 수 있다. /
================================================================================
Bayesian method:
- Calculate posterior probability
- by using prior probability and likelihood
- By using Bayesian method,
- you can get more information
- than the method where you use only likelihood
================================================================================
/ 이때 사전확률은 선험적으로 각 염기 위치(base position)가 유전변이가 /
/ 될 확률이며, 우도는 그 위치의 염기가 서로 다른 유전자형이 될 빈도(frequency)이다. /
================================================================================
prior probability
- probability of each base position becoming "genetic variation"
likelihood
- frequency of specific location's base becoming differening genetic type
================================================================================
/ 즉, 어느 /
/ locus에서 샘플 𝑖의 유전자형이 𝐺𝑖가 될 확률은 NGS 데이터 집합 𝐷와 베이즈 규칙(Bayes’ rule)에 /
/ 의하여 /
================================================================================
$$$p(G_i|D)
= \dfrac{p(G_i) p(D|G_i)}{ \sum\limits_{k=1}^{S} p(G_k) p(D|G_k)}$$$
$$$D$$$: NGS data set
$$$i$$$: ith sample from locus
$$$G_i$$$: genetic type of sample i
$$$S$$$: number of genetic type
$$$p(G_i)$$$: prior probability
$$$p(D|G_i)$$$: likelihood
- prior probability $$$p(G_i)$$$ is predicted
by using SNP database like dbSNP
================================================================================
/ 변이 추출 결과는 VCF(variant call format) [32] 형식의 파일에 저장된다. /
================================================================================
Result from variation calling (extracting genetic variant from read)
- is saved into VCF (variant call format) file
================================================================================
/ VCF 파일은 /
/ SNP/Indel로 확인된 유전변이 후보에 대한 정보, 즉 염색체 위치, 참조 염기, 치환/삽입/삭제 등이 /
/ 일어난 표본 염기, 변이 정확도(variation quality), 변이로 판명된 샘플의 빈도 등을 포함하고 있다. /
================================================================================
VCF file
- contains following information which is confirmed via SNP/Indel
- information about candidate genetic variation
- location of chromosome (of genetic variant occured?)
- reference sequence
- sample sequence which is occurred by
- substitution, insertion, deletion
- variation quality
- frequency of sample which is confirmed as genetic variation
================================================================================
/ 또한 변이 빈도(variation frequency)에 의해 SNP는 homozygous와 heterozygous 등 두 가지 /
/ 유형으로 나뉜다. Homozygous는 해당 위치에 정렬된 거의 모든 샘플에서 변이가 일어난 경우이고, /
/ heterozygous는 일부 샘플에서만 변이가 일어난 경우이다. 그림 5는 SAMtools를 사용하여 구한 /
/ VCF 형식의 파일이다. /
================================================================================
SNP:
- has following 2 types due to variation frequency
- homozygous
- heterozygous
================================================================================
/ Homozygous는 해당 위치에 정렬된 거의 모든 샘플에서 변이가 일어난 경우이고, /
/ heterozygous는 일부 샘플에서만 변이가 일어난 경우이다. 그림 5는 SAMtools를 사용하여 구한 /
/ VCF 형식의 파일이다.
================================================================================
Homozygous:
- Almost all samples which are aligned to reference sequence
have genetic variation
heterozygous:
- Only some part of samples which are aligned to reference sequence
have genetic variation
================================================================================
/ 그림 5는 SAMtools를 사용하여 구한 /
/ VCF 형식의 파일이다. /
================================================================================
VCF file example which is obtained via SAMtools
 ================================================================================
/ 변이 추출을 통해 얻어진 SNP/Indel은 잠재적인 것으로 모두 유의한 유전변이라고 간주하기 /
/ 어렵기 때문에 필터링을 통해 거짓양성(false-positive)에 해당하는 SNP/Indel을 제거하고 유의한 /
/ SNP/Indel만 골라내는 과정을 거친다. 보통 사용하는 방법은 Hardy-Weinberg 평형에서 벗어난 정도, /
/ 리드의 최대 및 최소 깊이, 인접한 Indels 등을 기준으로 조사한다. /
================================================================================
- You obtain SNP/Indel from "variation calling step"
- But you shouldn't consider all SNP/Indel as significant genetic variation
- So, you should filter false-positive SNP/Indel
- to have only significant SNP/Indel
- Method:
- Detect SNP/Indel which is out of Hardy-Weinberg equilibrium state
- Inspect maximal/minimal depth of reads
- Inspect neighboring Indels
- With them, you can classify significant SNL/Indel
================================================================================
/ 특별히 정해져 있는 기준이 없기 /
/ 때문에 정렬된 리드들의 깊이, 변이로 판명된 샘플의 빈도 등을 고려한 적정 기준을 세워 필터링을 /
/ 진행한다. 필터링을 위한 소프트웨어로 GATK의 VariantFiltration, vcfutil.pl, SAMtools, 그리고 /
/ VCFtools [32] 등이 있다. 특히 VCFtools은 VCF 파일을 다양한 방면으로 다루는 역할을 할 수 /
/ 있는데, 여러 파일을 합치거나 필요한 영역에 속하는 SNP만 추출할 수 있다. /
================================================================================
Software for filtering SNP/Indel
- GATK - VariantFiltration
- vcfutil.pl
- SAMtools
- VCFtools
================================================================================
/ NGS로 생성한 FASTQ 파일의 크기는 압축을 해도 샘플 당 기가 바이트(gigabyte, 109 bytes) /
/ 수준에 이른다. BAM/SAM 파일과 VCF 파일의 용량을 고려하면 샘플 100개의 파일 용량은 테라 /
/ 바이트(terabyte, 1012bytes) 수준이 된다.
================================================================================
FASTQ file (from NGS) size (after compression): GB per sample
FASTQ file + BAM/SAM + VCF file (from NGS): TB per 100 samples
================================================================================
/ Nvidia CUDA를 사용하여 BWA /
/ 알고리즘을 적용한 정렬 프로그램인 Barracuda /
================================================================================
BWA algorith with CUDA: Barracuda
================================================================================
/ 1990년대까지 개인용 컴퓨터의 메모리(random access memory, RAM)는 주로 32-bits /
/ 운영체제에서 사용되었기 때문에 용량은 최대 약 4GB까지 허용되었다. 따라서 운영체제 및 /
/ 실행중인 기타 소프트웨어를 위한 메모리를 포함하여 4GB 이상의 메모리를 요구하는 유전체 /
/ 데이터의 분석은 불가능하였다. 2004년경 64-bits 컴퓨팅을 지원하는 CPU가 등장함에 따라 운영체제 /
/ 역시 최대 약 8TB의 메모리를 지원할 수 있기 되어, NGS 데이터 전체를 메모리에 탑재하여 분석할 /
/ 수 있게 되었다 /
32 bit OS allows 4GB maximal memory
64 bit OS allows 8TB maximal memory
================================================================================
/ NGS로 대용량의 유전정보를 생산할 수 있음에 따라 인간뿐만 아니라 다양한 동식물의 /
/ 유전체 해독이 보편화 되고 있으며, 질병 진단 및 예측과 유용 유전정보 발굴 및 육종에 응용 등이 /
/ 가능하게 되었다. NGS 데이터의 재분석을 통한 생물학적 해석과 활용은 원시 데이터의 분석 결과인 /
/ SNP/Indel calling의 정확도가 전제되어야 하며, 정확도는 분석 방법론과 알고리즘에 영향을 받는다. /
================================================================================
NGS
- can create massive genetic information data
Re-analysis on NGS data
- can be used for biological interpretation
- but integrity of "SNP/Indel calling" should be prepared for re-analysis on NGS data
================================================================================
/ 따라서 FASTQ 파일에서 SNP calling에 이르는 분석 파이프라인에 대한 지속적인 연구가 필요하며, /
/ 생물학적 연구 목적에 따라 기능 추가/삭제 등을 포함한 파이프라인의 구조를 변화시킨 맞춤형 /
/ 파이프라인을 구축할 수 있는 데이터 분석 기술이 요구된다. /
================================================================================
Researches are needed for pipeline
- FASTQ file
...
- SNP calling
================================================================================
/ 현재는 1,000 genome /
/ project와 같은 대형 유전체 프로젝트에서 사용하는 파이프라인을 표준으로 간주하고 있으나, 아직 /
/ 의견 일치를 보이는 것은 아니며, 또한 파이프라인이 워낙 복잡하여 ‘표준 파이프라인’이 가능한지도 /
/ 현재로는 의문이다. /
================================================================================
Pipeline which is used for "1,000 genome project" is considered as standard
================================================================================
/ 한편 “The Genome in a Bottle Consortium” [37, 38]에서 DNA 시퀀싱의 표준 /
/ 데이터를 제공하고 있음으로, 향 후 다양한 소프트웨어 및 파이프라인을 비교 검토하는데 사용할 수 /
/ 있을 것으로 기대된다 /
================================================================================
The Genome in a Bottle Consortium provides standard data for DNA sequencing
================================================================================
================================================================================
/ 변이 추출을 통해 얻어진 SNP/Indel은 잠재적인 것으로 모두 유의한 유전변이라고 간주하기 /
/ 어렵기 때문에 필터링을 통해 거짓양성(false-positive)에 해당하는 SNP/Indel을 제거하고 유의한 /
/ SNP/Indel만 골라내는 과정을 거친다. 보통 사용하는 방법은 Hardy-Weinberg 평형에서 벗어난 정도, /
/ 리드의 최대 및 최소 깊이, 인접한 Indels 등을 기준으로 조사한다. /
================================================================================
- You obtain SNP/Indel from "variation calling step"
- But you shouldn't consider all SNP/Indel as significant genetic variation
- So, you should filter false-positive SNP/Indel
- to have only significant SNP/Indel
- Method:
- Detect SNP/Indel which is out of Hardy-Weinberg equilibrium state
- Inspect maximal/minimal depth of reads
- Inspect neighboring Indels
- With them, you can classify significant SNL/Indel
================================================================================
/ 특별히 정해져 있는 기준이 없기 /
/ 때문에 정렬된 리드들의 깊이, 변이로 판명된 샘플의 빈도 등을 고려한 적정 기준을 세워 필터링을 /
/ 진행한다. 필터링을 위한 소프트웨어로 GATK의 VariantFiltration, vcfutil.pl, SAMtools, 그리고 /
/ VCFtools [32] 등이 있다. 특히 VCFtools은 VCF 파일을 다양한 방면으로 다루는 역할을 할 수 /
/ 있는데, 여러 파일을 합치거나 필요한 영역에 속하는 SNP만 추출할 수 있다. /
================================================================================
Software for filtering SNP/Indel
- GATK - VariantFiltration
- vcfutil.pl
- SAMtools
- VCFtools
================================================================================
/ NGS로 생성한 FASTQ 파일의 크기는 압축을 해도 샘플 당 기가 바이트(gigabyte, 109 bytes) /
/ 수준에 이른다. BAM/SAM 파일과 VCF 파일의 용량을 고려하면 샘플 100개의 파일 용량은 테라 /
/ 바이트(terabyte, 1012bytes) 수준이 된다.
================================================================================
FASTQ file (from NGS) size (after compression): GB per sample
FASTQ file + BAM/SAM + VCF file (from NGS): TB per 100 samples
================================================================================
/ Nvidia CUDA를 사용하여 BWA /
/ 알고리즘을 적용한 정렬 프로그램인 Barracuda /
================================================================================
BWA algorith with CUDA: Barracuda
================================================================================
/ 1990년대까지 개인용 컴퓨터의 메모리(random access memory, RAM)는 주로 32-bits /
/ 운영체제에서 사용되었기 때문에 용량은 최대 약 4GB까지 허용되었다. 따라서 운영체제 및 /
/ 실행중인 기타 소프트웨어를 위한 메모리를 포함하여 4GB 이상의 메모리를 요구하는 유전체 /
/ 데이터의 분석은 불가능하였다. 2004년경 64-bits 컴퓨팅을 지원하는 CPU가 등장함에 따라 운영체제 /
/ 역시 최대 약 8TB의 메모리를 지원할 수 있기 되어, NGS 데이터 전체를 메모리에 탑재하여 분석할 /
/ 수 있게 되었다 /
32 bit OS allows 4GB maximal memory
64 bit OS allows 8TB maximal memory
================================================================================
/ NGS로 대용량의 유전정보를 생산할 수 있음에 따라 인간뿐만 아니라 다양한 동식물의 /
/ 유전체 해독이 보편화 되고 있으며, 질병 진단 및 예측과 유용 유전정보 발굴 및 육종에 응용 등이 /
/ 가능하게 되었다. NGS 데이터의 재분석을 통한 생물학적 해석과 활용은 원시 데이터의 분석 결과인 /
/ SNP/Indel calling의 정확도가 전제되어야 하며, 정확도는 분석 방법론과 알고리즘에 영향을 받는다. /
================================================================================
NGS
- can create massive genetic information data
Re-analysis on NGS data
- can be used for biological interpretation
- but integrity of "SNP/Indel calling" should be prepared for re-analysis on NGS data
================================================================================
/ 따라서 FASTQ 파일에서 SNP calling에 이르는 분석 파이프라인에 대한 지속적인 연구가 필요하며, /
/ 생물학적 연구 목적에 따라 기능 추가/삭제 등을 포함한 파이프라인의 구조를 변화시킨 맞춤형 /
/ 파이프라인을 구축할 수 있는 데이터 분석 기술이 요구된다. /
================================================================================
Researches are needed for pipeline
- FASTQ file
...
- SNP calling
================================================================================
/ 현재는 1,000 genome /
/ project와 같은 대형 유전체 프로젝트에서 사용하는 파이프라인을 표준으로 간주하고 있으나, 아직 /
/ 의견 일치를 보이는 것은 아니며, 또한 파이프라인이 워낙 복잡하여 ‘표준 파이프라인’이 가능한지도 /
/ 현재로는 의문이다. /
================================================================================
Pipeline which is used for "1,000 genome project" is considered as standard
================================================================================
/ 한편 “The Genome in a Bottle Consortium” [37, 38]에서 DNA 시퀀싱의 표준 /
/ 데이터를 제공하고 있음으로, 향 후 다양한 소프트웨어 및 파이프라인을 비교 검토하는데 사용할 수 /
/ 있을 것으로 기대된다 /
================================================================================
The Genome in a Bottle Consortium provides standard data for DNA sequencing
================================================================================