https://www.youtube.com/watch?v=YkDoA3lZb5k&list=PLaE61CK5r6_l2fxVp3r3OP0fgTSTdQUoQ
================================================================================
- Read FASTA format file (title + sequence)
- Read sequence
- Calculate GC content
python reverse_complement.py > input.fast
# 0.456
================================================================================
import unittest
import sys
class Fasta:
def __init__(self,title,sequence):
self.title=title
self.sequence=sequence
def get_gc_content(self):
gc=self.sequence.count("G")+self.sequence.count("C")
return gc/float(len(self.sequence))
================================================================================
class FastaTest(unittest.TestCase)::
def test_fasta_object(self):
my_fast=Fasta("test1","AGTC")
self.assertEqual(my_fasta.title,"test1")
self.assertEqual(my_fasta.sequence,"AGTC")
def test_gc_content(self):
my_fast=Fasta("test1","AGTC")
self.assertEqual(my_fasta.get_gc_content(),0.5)
================================================================================
if __name__=="__main__":
# unittest.main()
================================================================================
sequence=[]
for line in sys.stdin:
line=line.strip()
if line.startswith(">"):
title=line[1:]
else:
sequence.append(line)
sequence="".join(sequence)
my_fasta=Fasta(title,sequence)
print(my_fasta.get_gc_content())
================================================================================
cat test.fasta
 ================================================================================
python rc2.py < test.fasta
================================================================================
Exercise
- Read DNA sequence from one FASTA file
- Print "reverse complement"
python reverse_complement.py<input.fasta
================================================================================
python rc2.py < test.fasta
================================================================================
Exercise
- Read DNA sequence from one FASTA file
- Print "reverse complement"
python reverse_complement.py<input.fasta
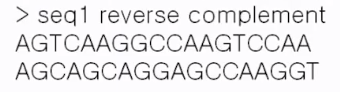 ================================================================================
import unittest
import sys
class Fasta:
def __init__(self,title,sequence):
self.title=title
self.sequence=sequence
def get_gc_content(self):
gc=self.sequence.count("G")+self.sequence.count("C")
return gc/float(len(self.sequence))
def reverse_complement(self):
base_pairs={
"A":"T",
"T":"A",
"G":"C",
"C":"G"}
result=[]
for nucleotide in self.sequence:
result.append(base_pairs[nucleotide])
return ''.join(reverse(result))
================================================================================
class FastaTest(unittest.TestCase)::
def test_fasta_object(self):
my_fast=Fasta("test1","AGTC")
self.assertEqual(my_fasta.title,"test1")
self.assertEqual(my_fasta.sequence,"AGTC")
def test_gc_content(self):
my_fast=Fasta("test1","AGTC")
self.assertEqual(my_fasta.get_gc_content(),0.5)
def test_reverse_complement(self):
my_fast=Fasta("test1","AGTC")
# "ATGC"
self.assertEqual(my_fasta.reverse_complement(),"GACT")
================================================================================
def main():
sequence=[]
for line in sys.stdin:
line=line.strip()
if line.startswith(">"):
title=line[1:]
else:
sequence.append(line)
sequence="".join(sequence)
my_fasta=Fasta(title,sequence)
print(my_fasta.get_gc_content())
if __name__=="__main__":
unittest.main()
# main()
================================================================================
import unittest
import sys
class Fasta:
def __init__(self,title,sequence):
self.title=title
self.sequence=sequence
def get_gc_content(self):
gc=self.sequence.count("G")+self.sequence.count("C")
return gc/float(len(self.sequence))
def reverse_complement(self):
base_pairs={
"A":"T",
"T":"A",
"G":"C",
"C":"G"}
result=[]
for nucleotide in self.sequence:
result.append(base_pairs[nucleotide])
return ''.join(reverse(result))
================================================================================
class FastaTest(unittest.TestCase)::
def test_fasta_object(self):
my_fast=Fasta("test1","AGTC")
self.assertEqual(my_fasta.title,"test1")
self.assertEqual(my_fasta.sequence,"AGTC")
def test_gc_content(self):
my_fast=Fasta("test1","AGTC")
self.assertEqual(my_fasta.get_gc_content(),0.5)
def test_reverse_complement(self):
my_fast=Fasta("test1","AGTC")
# "ATGC"
self.assertEqual(my_fasta.reverse_complement(),"GACT")
================================================================================
def main():
sequence=[]
for line in sys.stdin:
line=line.strip()
if line.startswith(">"):
title=line[1:]
else:
sequence.append(line)
sequence="".join(sequence)
my_fasta=Fasta(title,sequence)
print(my_fasta.get_gc_content())
if __name__=="__main__":
unittest.main()
# main()