https://www.youtube.com/watch?v=-0Xur_HtHaM&list=PLaE61CK5r6_l2fxVp3r3OP0fgTSTdQUoQ
================================================================================
from Bio.Seq import Seq
from Bio.Alphabet import IUPAC
# c a: default sequence string
a=Seq("AGTCAGTC")
print("a",a)
 ================================================================================
# c a: DNA sequence string
a=Seq("AGTCAGTC",IUPAC.unambiguous_dna)
print("a",a)
================================================================================
# c a: DNA sequence string
a=Seq("AGTCAGTC",IUPAC.unambiguous_dna)
print("a",a)
 a.complement
a.complement
 a.reverse_complement
a.reverse_complement
 ================================================================================
================================================================================
 ================================================================================
================================================================================
 IUPAC: institute which defines "alphabets"
================================================================================
IUPAC: definition for 20 amino acids (in form of alphabet)
ExtendedIUPACProtein:
some amino acids are added in alphabet form
IUPAC: institute which defines "alphabets"
================================================================================
IUPAC: definition for 20 amino acids (in form of alphabet)
ExtendedIUPACProtein:
some amino acids are added in alphabet form
 ================================================================================
For nucleotide,
- IUPACUnambiguousDNA
- IUPACAmbiguousDNA:
one alphabet which indicates over-2 bases, for example, alphabet_1 indicates to both A and G
- ExtendedIUPACDNA
- B: 5-bromouridine
- D: 5,6-dihydrouridine
- S: Thiouridine
- W: wyosine
- IUPACAmbiguousDNA
- IUPACAmbiguousRNA
================================================================================
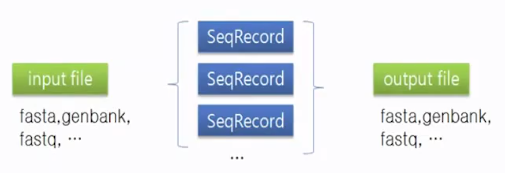
SeqRecord object
Attributes
- .seq: Seq object
- .id: ID which is used to identify the sequence
- .name: title string
- .description: human readable description
- .letter_annotations: dict()
- .annotations: dict()
- .features: list of SeqFeature objects
- .dbxrefs: list of database cross-reference
================================================================================
from Bio.Seq import Seq
from Bio.SeqRecord import SeqRecord
simple_seq=Seq("AGTC")
simple_seq_r=SeqRecord(simple_seq)
================================================================================
SeqFeature object
Attributes
- .type: CDS, gene
- .location
- .qualifiers: dict(), additional information
- .sub_features: list of sub SeqFeatures objects
================================================================================
For nucleotide,
- IUPACUnambiguousDNA
- IUPACAmbiguousDNA:
one alphabet which indicates over-2 bases, for example, alphabet_1 indicates to both A and G
- ExtendedIUPACDNA
- B: 5-bromouridine
- D: 5,6-dihydrouridine
- S: Thiouridine
- W: wyosine
- IUPACAmbiguousDNA
- IUPACAmbiguousRNA
================================================================================
SeqRecord object
Attributes
- .seq: Seq object
- .id: ID which is used to identify the sequence
- .name: title string
- .description: human readable description
- .letter_annotations: dict()
- .annotations: dict()
- .features: list of SeqFeature objects
- .dbxrefs: list of database cross-reference
================================================================================
from Bio.Seq import Seq
from Bio.SeqRecord import SeqRecord
simple_seq=Seq("AGTC")
simple_seq_r=SeqRecord(simple_seq)
================================================================================
SeqFeature object
Attributes
- .type: CDS, gene
- .location
- .qualifiers: dict(), additional information
- .sub_features: list of sub SeqFeatures objects
 ================================================================================
================================================================================
 Download file
================================================================================
Download file
================================================================================
 ================================================================================
SeqIO
- For sequence input and output
================================================================================
SeqIO
- For sequence input and output
 from Bio import SeqIO
record_iterator=SeqIO.parse("input.gb","gb")
for record in record_iterator:
print(record.format("fasta"))
# Or you can also use
# SeqIO.write(record,"outputfilename","fasta")
================================================================================
gi=SeqIO.parse("sequence.gb","gb")
from Bio import SeqIO
record_iterator=SeqIO.parse("input.gb","gb")
for record in record_iterator:
print(record.format("fasta"))
# Or you can also use
# SeqIO.write(record,"outputfilename","fasta")
================================================================================
gi=SeqIO.parse("sequence.gb","gb")
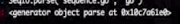 record=gi.next()
record=gi.next()
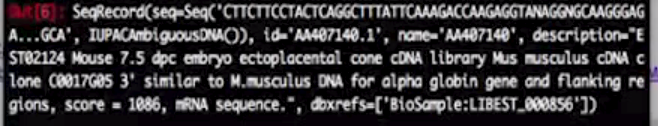 record.id
record.id
 record.name
record.name
 record.description
record.description
 record.annotations
record.annotations
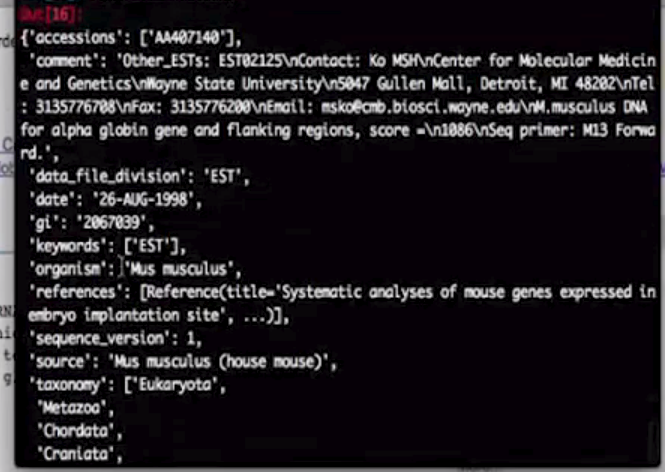 f=record.features
f=record.features
 f.type
# source
f.location
f.type
# source
f.location
 ================================================================================
print('record.format("gb")',record.format("gb"))
# gene bank format
print('record.format("fasta")',record.format("fasta"))
# fasta format
print('record.format("embl")',record.format("embl"))
# embl format
================================================================================
Formats which you can use
================================================================================
print('record.format("gb")',record.format("gb"))
# gene bank format
print('record.format("fasta")',record.format("fasta"))
# fasta format
print('record.format("embl")',record.format("embl"))
# embl format
================================================================================
Formats which you can use
 ================================================================================
Quiz
- Read multiple FASTA files
- Write reverse complement in FASTA format
import sys
from Bio import SeqIO
for record in SeqIO.parse(sys.stdin,"fasta"):
record.seq=record.seq.reverse_complement()
record.description=record.description+" reverse complemented"
print(record.format("fasta"))
# print(record.format("gb "))
# Error: fasta ---> gb can't be possible
# because fasta has some data which gb doesn't have
================================================================================
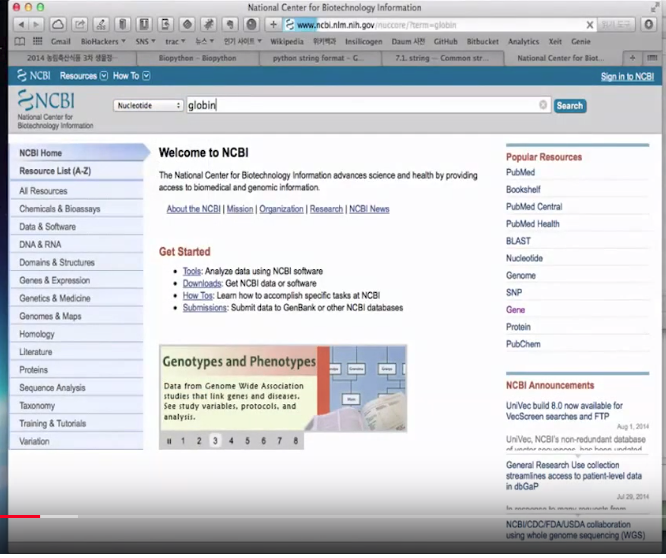
How to use Entrez (which is the name of NCBI database)
BioPython's Entrez is for getting SeqRecord (data) from NCBI
from Bio import Entrez
Entrez.email="yours@yours.com"
print(Entrez.einfo().read())
================================================================================
Quiz
- Read multiple FASTA files
- Write reverse complement in FASTA format
import sys
from Bio import SeqIO
for record in SeqIO.parse(sys.stdin,"fasta"):
record.seq=record.seq.reverse_complement()
record.description=record.description+" reverse complemented"
print(record.format("fasta"))
# print(record.format("gb "))
# Error: fasta ---> gb can't be possible
# because fasta has some data which gb doesn't have
================================================================================
How to use Entrez (which is the name of NCBI database)
BioPython's Entrez is for getting SeqRecord (data) from NCBI
from Bio import Entrez
Entrez.email="yours@yours.com"
print(Entrez.einfo().read())
 All kinds of DB of Entrez, which you can use
================================================================================
from Bio import Entrez
Entrez.email="yours@yours.com"
handle=Entrez.esearch(db="pubmed",term="biopython")
record=Entrez.read(handle)
All kinds of DB of Entrez, which you can use
================================================================================
from Bio import Entrez
Entrez.email="yours@yours.com"
handle=Entrez.esearch(db="pubmed",term="biopython")
record=Entrez.read(handle)
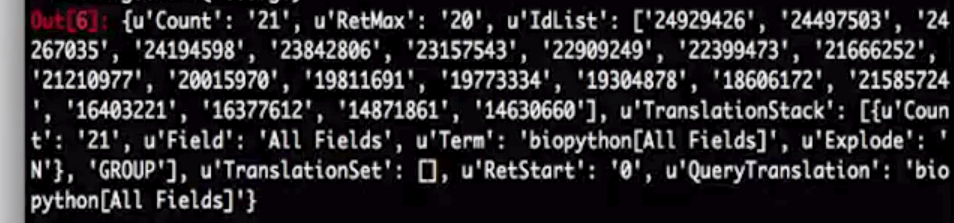 # PubMed's IDs
# Each one is thesis paper
record["IdList"]
# PubMed's IDs
# Each one is thesis paper
record["IdList"]
 ================================================================================
# Organism: Cypripedioidease
# Gene name: matK
handle=Entrez.esearch(db="nucleotide",term="Cypripedioidease[Organ] AND matK[Gene]")
record=Entrez.read(handle)
record["Count"]
record["IdList"]
================================================================================
from Bio import Entrez
Entrez.email="yours@yours.com"
handle=Entrez.efectch(db="nucleotide",id="186972394",rettype="gb",retmode="text")
record=SeqIO.read(handle,"genbank")
================================================================================
from Bio import Entrez
Entrez.email="yours@yours.com"
handle=Entrez.esearch(db="pubmed",term="biopython")
record=Entrez.read(handle)
record["IdList"]
================================================================================
# Organism: Cypripedioidease
# Gene name: matK
handle=Entrez.esearch(db="nucleotide",term="Cypripedioidease[Organ] AND matK[Gene]")
record=Entrez.read(handle)
record["Count"]
record["IdList"]
================================================================================
from Bio import Entrez
Entrez.email="yours@yours.com"
handle=Entrez.efectch(db="nucleotide",id="186972394",rettype="gb",retmode="text")
record=SeqIO.read(handle,"genbank")
================================================================================
from Bio import Entrez
Entrez.email="yours@yours.com"
handle=Entrez.esearch(db="pubmed",term="biopython")
record=Entrez.read(handle)
record["IdList"]
 idlist=record["IdList"]
handle=Entrez.efectch(db="pubmed",id=idlist,rettype="medline",retmode="text")
records=Medline.parse(handle)
for record in records::
print(record["AU"])
idlist=record["IdList"]
handle=Entrez.efectch(db="pubmed",id=idlist,rettype="medline",retmode="text")
records=Medline.parse(handle)
for record in records::
print(record["AU"])
