https://www.youtube.com/watch?v=TdAA1MFSjog&list=PLaE61CK5r6_l2fxVp3r3OP0fgTSTdQUoQ
================================================================================
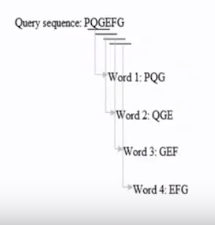
BLAST: Basic Local Alignment Search Tool
Biologically homology search for protein and necleotide
- blastn: for nucleotide
- blastp (blastx): for protein (6 possible translations)
================================================================================
 ================================================================================
================================================================================
 ================================================================================
Result from BLAST
================================================================================
Result from BLAST
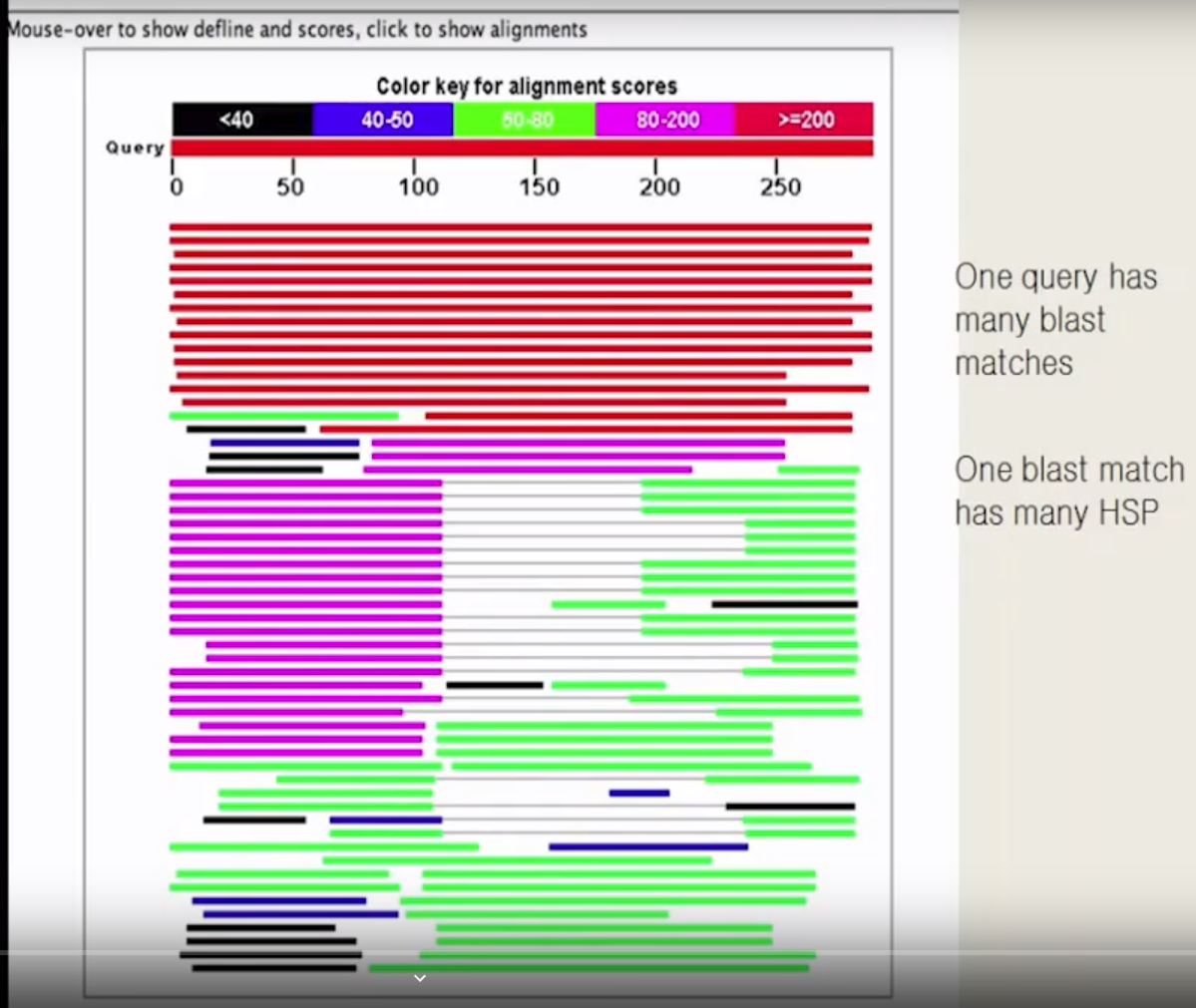 Length of query (protein or nucleotide) sequence: around 270 bp
- HSP: high score position
================================================================================
Install BLAST
sudo apt-get install ncbi-blast+
# Download data file
wget ftp://ftp.ncbi.nih.gov/blast/db/FASTA/swissprot.gz
# Uncompress data file
gunzip swissprot.gz
# Create data format which can be consumed in BLAST
makeblastdb -in swissprot -dbtype prot
# outfmt: output format is 7th one
# query.fasta: input file
# query.blout: output file
blastx -db swissprot -outfmt 7 < query.fasta > query.blout
================================================================================
Length of query (protein or nucleotide) sequence: around 270 bp
- HSP: high score position
================================================================================
Install BLAST
sudo apt-get install ncbi-blast+
# Download data file
wget ftp://ftp.ncbi.nih.gov/blast/db/FASTA/swissprot.gz
# Uncompress data file
gunzip swissprot.gz
# Create data format which can be consumed in BLAST
makeblastdb -in swissprot -dbtype prot
# outfmt: output format is 7th one
# query.fasta: input file
# query.blout: output file
blastx -db swissprot -outfmt 7 < query.fasta > query.blout
================================================================================
 Formatting options
- 0: traditional format, showing all alignments
- 5,6,7: much used
================================================================================
Parse output file
import sys
from Bio.Blast import NCBIStandalone
blast_parser=NCBIStandalone.BlastParser()
for record in NCBIStandalone.Iterator(sys.stdin,blast_parser):
for alignment in record.alignments:
for hsp in alignment.hsps:
print(alignment.title)
print(alignment.length)
print(alignment.expect)
print(alignment.query)
print(alignment.match)
print(alignment.sbjct)
================================================================================
Formatting options
- 0: traditional format, showing all alignments
- 5,6,7: much used
================================================================================
Parse output file
import sys
from Bio.Blast import NCBIStandalone
blast_parser=NCBIStandalone.BlastParser()
for record in NCBIStandalone.Iterator(sys.stdin,blast_parser):
for alignment in record.alignments:
for hsp in alignment.hsps:
print(alignment.title)
print(alignment.length)
print(alignment.expect)
print(alignment.query)
print(alignment.match)
print(alignment.sbjct)
================================================================================
 Result from BLAST
Result from BLAST
 ================================================================================
import sys
from Bio.Blast import NCBIXML
for record in NCBIXML.parse(sys.stdin):
for alignment in record.alignments:
for hsp in alignment.hsps:
print(alignment.title)
print(alignment.length)
print(alignment.expect)
print(alignment.query)
print(alignment.match)
print(alignment.sbjct)
================================================================================
Quiz
- You obtain result from BLAST
- You insert that result into pipeline
- You shuld get summrization of BLAST result
- Contraint: expected_value<0.005
- Run example
python blast_summary.py 0.005 < my_quesry.blout.pairwise
- Summarization example:
================================================================================
import sys
from Bio.Blast import NCBIXML
for record in NCBIXML.parse(sys.stdin):
for alignment in record.alignments:
for hsp in alignment.hsps:
print(alignment.title)
print(alignment.length)
print(alignment.expect)
print(alignment.query)
print(alignment.match)
print(alignment.sbjct)
================================================================================
Quiz
- You obtain result from BLAST
- You insert that result into pipeline
- You shuld get summrization of BLAST result
- Contraint: expected_value<0.005
- Run example
python blast_summary.py 0.005 < my_quesry.blout.pairwise
- Summarization example:
 ================================================================================
Subprocess
Subprocess Python module allows you to spawn new processes,
you to connect to their input/output/error pipes,
and you can obtain their return codes
from subprocess import Popen,PIPE
# Create new process, run command, prepare "pipe" to store result text
p=Popen(['ls','-la'],stdout=PIPE)
# Get result string from process
output=p.communicate()[0]
================================================================================
ps -ef | grep zsh
result of "ps -ef" becomes "input" to "grep zsh"
================================================================================
p1=Popen(['ls','-al'],stdout=PIPE)
p2=Popen(['grep','zsh'],stdin=p1.stdout,stdout=PIPE)
p1.stdout.close()
output=p2.communicate()[0]
================================================================================
from Bio import SeqIO
from subprocess import Popen,PIPE
my_seq_record=SeqIO.parse("query.fasta","fasta")
input_query=my_seq_record.format("fasta")
p=Popen(["blastx","-db","swissprot","-outfmt","6"],stdin=input_query)
output=p.communicate()[0]
number_of_hsps=len(output.splitlines())
================================================================================
Quiz
- You can search data in nucleotide_NCBI by using "specific given keyword from user"
- You obtain 10 number of data
- You search above data from swissprot_DB by using blastx
- You should answer how many number of proteins are
- Constraint: E-value < $$$10^{-5}$$$, duplicated number of protein is not allowed
- Run example:
python count-concerned-proteins.py "apoptosis"
# 356
================================================================================
Implementation
vi search.py
import sys
from Bio import Entrez
from Bio import SeqIO
from subprocess import Popen,PIPE
from Bio.Blast import NCBIStandalone
StringIO
================================================================================
keyword=sys.argv[1]
handle=Entrez.esearch(db="nucleotide",term=keyword)
result=Entrez.read(handle)
idlist=result["IdList"]
================================================================================
blast_parser=NCBIStandalone.BlastParser()
================================================================================
handle=Entrez.efetch(db="nucleotide",id=idlist,rettype="gb",retmode="text")
for record in SeqIO.parse(hanlde,"gb"):
# print(record.format("fasta"))
input=record.format("fasta")
p=Popen(['blastx','-db','swissprot','-outfmt','0'],stdin=input)
output=p.communicate()[0]
================================================================================
total=0
for result in NCBIStandalone.Iterator(StringIO(output),blast_parser):
for alignment in result.alignments:
first_hsp=alignmentts.hsps[0]
if first_hsp<0.0005:
total+=len(result.alignments)
================================================================================
# Run
python search.py apoptosis
================================================================================
Subprocess
Subprocess Python module allows you to spawn new processes,
you to connect to their input/output/error pipes,
and you can obtain their return codes
from subprocess import Popen,PIPE
# Create new process, run command, prepare "pipe" to store result text
p=Popen(['ls','-la'],stdout=PIPE)
# Get result string from process
output=p.communicate()[0]
================================================================================
ps -ef | grep zsh
result of "ps -ef" becomes "input" to "grep zsh"
================================================================================
p1=Popen(['ls','-al'],stdout=PIPE)
p2=Popen(['grep','zsh'],stdin=p1.stdout,stdout=PIPE)
p1.stdout.close()
output=p2.communicate()[0]
================================================================================
from Bio import SeqIO
from subprocess import Popen,PIPE
my_seq_record=SeqIO.parse("query.fasta","fasta")
input_query=my_seq_record.format("fasta")
p=Popen(["blastx","-db","swissprot","-outfmt","6"],stdin=input_query)
output=p.communicate()[0]
number_of_hsps=len(output.splitlines())
================================================================================
Quiz
- You can search data in nucleotide_NCBI by using "specific given keyword from user"
- You obtain 10 number of data
- You search above data from swissprot_DB by using blastx
- You should answer how many number of proteins are
- Constraint: E-value < $$$10^{-5}$$$, duplicated number of protein is not allowed
- Run example:
python count-concerned-proteins.py "apoptosis"
# 356
================================================================================
Implementation
vi search.py
import sys
from Bio import Entrez
from Bio import SeqIO
from subprocess import Popen,PIPE
from Bio.Blast import NCBIStandalone
StringIO
================================================================================
keyword=sys.argv[1]
handle=Entrez.esearch(db="nucleotide",term=keyword)
result=Entrez.read(handle)
idlist=result["IdList"]
================================================================================
blast_parser=NCBIStandalone.BlastParser()
================================================================================
handle=Entrez.efetch(db="nucleotide",id=idlist,rettype="gb",retmode="text")
for record in SeqIO.parse(hanlde,"gb"):
# print(record.format("fasta"))
input=record.format("fasta")
p=Popen(['blastx','-db','swissprot','-outfmt','0'],stdin=input)
output=p.communicate()[0]
================================================================================
total=0
for result in NCBIStandalone.Iterator(StringIO(output),blast_parser):
for alignment in result.alignments:
first_hsp=alignmentts.hsps[0]
if first_hsp<0.0005:
total+=len(result.alignments)
================================================================================
# Run
python search.py apoptosis