https://www.youtube.com/watch?v=y42KBKvanRs&list=PLaE61CK5r6_l2fxVp3r3OP0fgTSTdQUoQ&index=2&t=727s
================================================================================
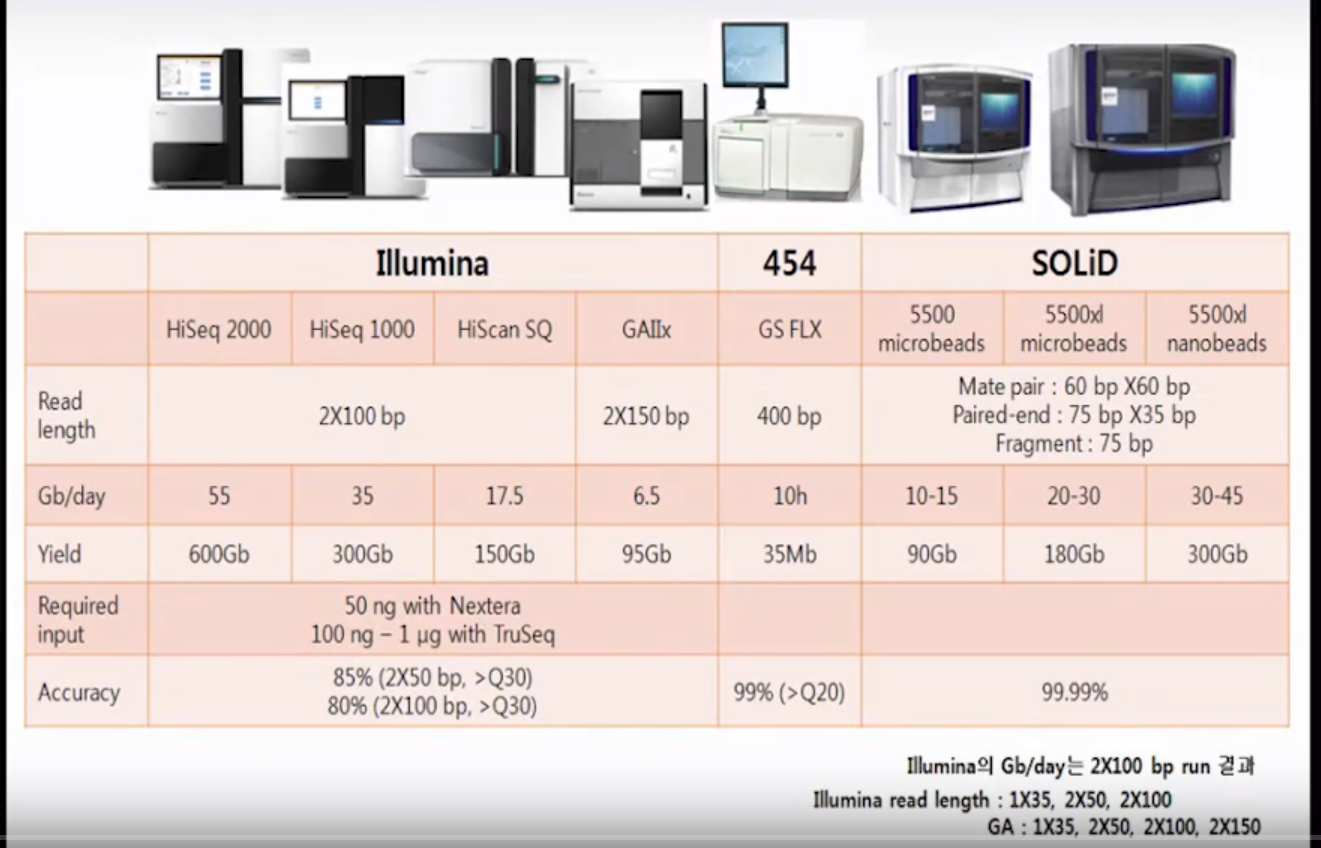
NGS sequencers
 ================================================================================
Application of NGS data
================================================================================
Application of NGS data
 ================================================================================
A high-level overview of NGS data processing
================================================================================
A high-level overview of NGS data processing
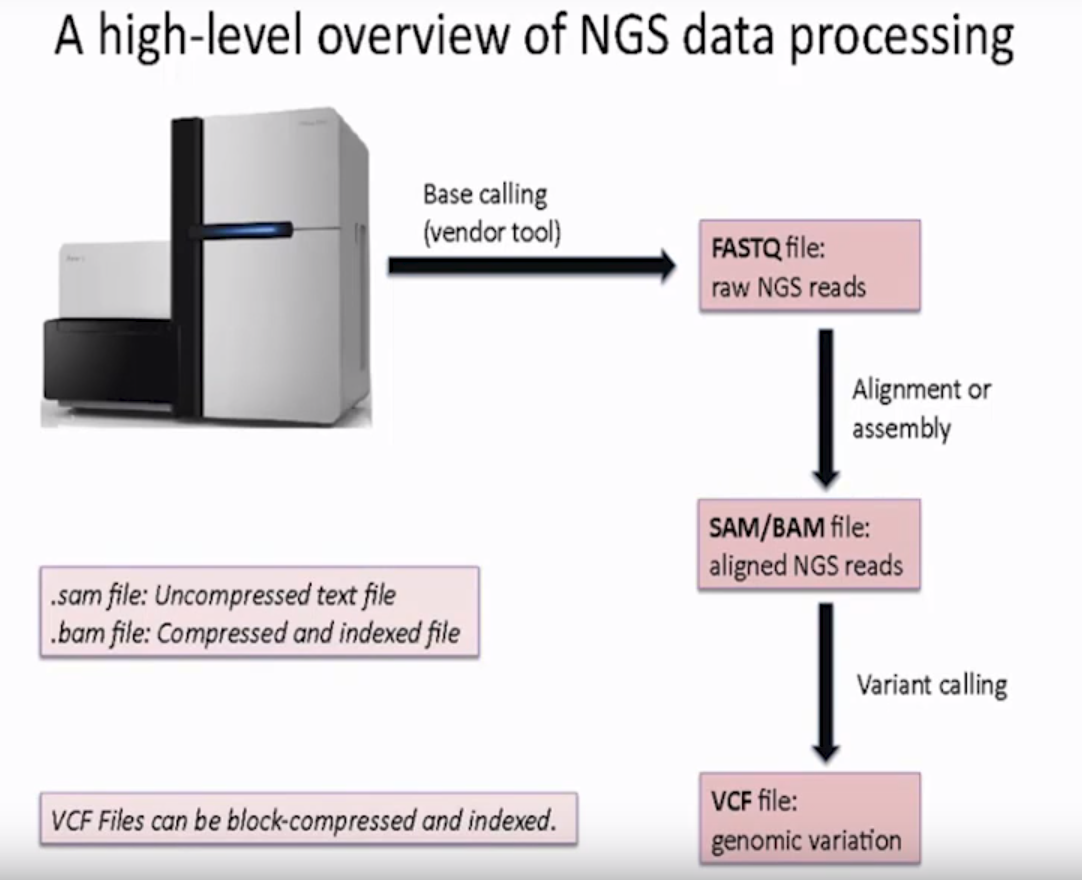 - Base calling
- Get FASTQ file
- Perform alignment or assembly
- Get SAM/BAM file
- Perform variant calling
- Get VCF file
================================================================================
Scope and schema of the Best practices
- Base calling
- Get FASTQ file
- Perform alignment or assembly
- Get SAM/BAM file
- Perform variant calling
- Get VCF file
================================================================================
Scope and schema of the Best practices
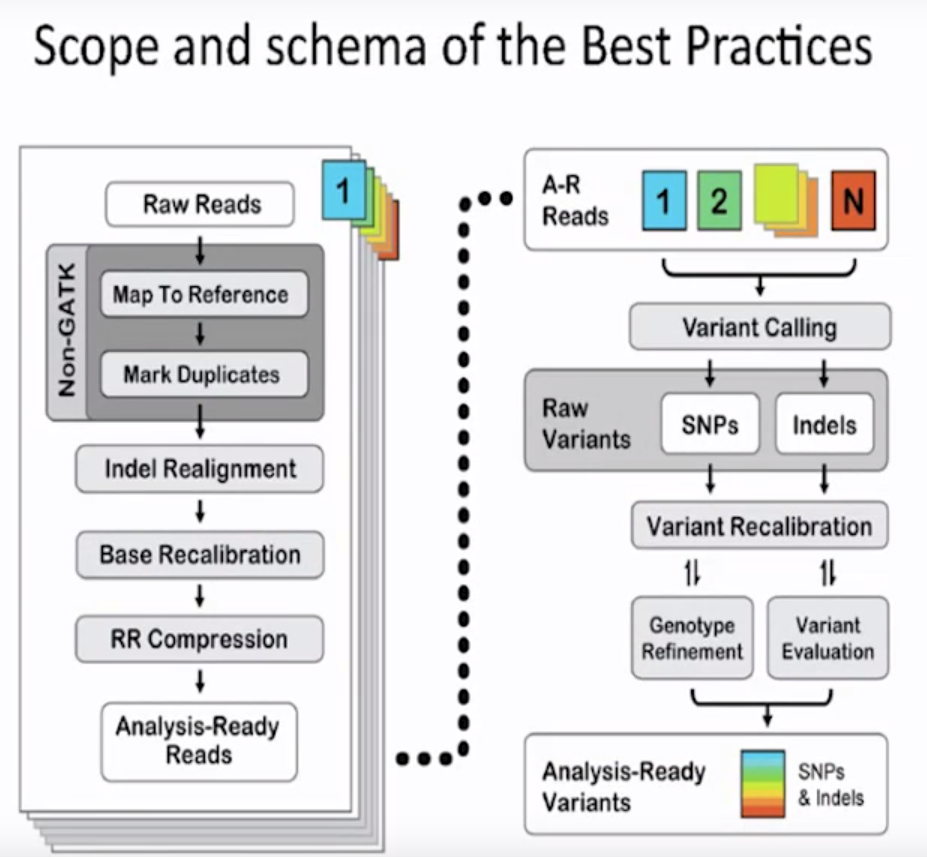 - If you analyze 100 number of cancer patients, sample becomes 100 number of ones
- You can see 100 number of samples in the left
- Non-GATK: they can't be performed by GATK
================================================================================
FASTQ: raw unaligned reads
1 recors takes 4 lines
1th line: identifier, title, sequence name (read's name, group, etc)
2nd line: sequence data (short in the case of NGS)
3rd line: + (optional, sequence name again)
4th line: associated quality score
- If you analyze 100 number of cancer patients, sample becomes 100 number of ones
- You can see 100 number of samples in the left
- Non-GATK: they can't be performed by GATK
================================================================================
FASTQ: raw unaligned reads
1 recors takes 4 lines
1th line: identifier, title, sequence name (read's name, group, etc)
2nd line: sequence data (short in the case of NGS)
3rd line: + (optional, sequence name again)
4th line: associated quality score
 ================================================================================
================================================================================
 - How to read quality line is different per seqeuncers
- How to read quality line is different per seqeuncers
 ================================================================================
from Bio import SeqIO
for record in SeqIO.parse("test1.fq","fastq"):
print(record.format("qual"))
print(record.letter_annotations)
record.format("fastq-sanger") # This is same format as fastq
record.format("fastq-solexa")
record.format("fastq-illumina")
================================================================================
from Bio import SeqIO
g=SeqIO.parse("sample_1.fq","fastq")
record=g.next()
================================================================================
from Bio import SeqIO
for record in SeqIO.parse("test1.fq","fastq"):
print(record.format("qual"))
print(record.letter_annotations)
record.format("fastq-sanger") # This is same format as fastq
record.format("fastq-solexa")
record.format("fastq-illumina")
================================================================================
from Bio import SeqIO
g=SeqIO.parse("sample_1.fq","fastq")
record=g.next()
 print(record.format("fasta"))
print(record.format("fasta"))
 print(record.format("fastq"))
print(record.format("fastq"))
 record.letter_annotations
record.format("qual")
record.letter_annotations
record.format("qual")
 ================================================================================
The BAM format stores aligned reads and is technology independent
================================================================================
The BAM format stores aligned reads and is technology independent
 ================================================================================
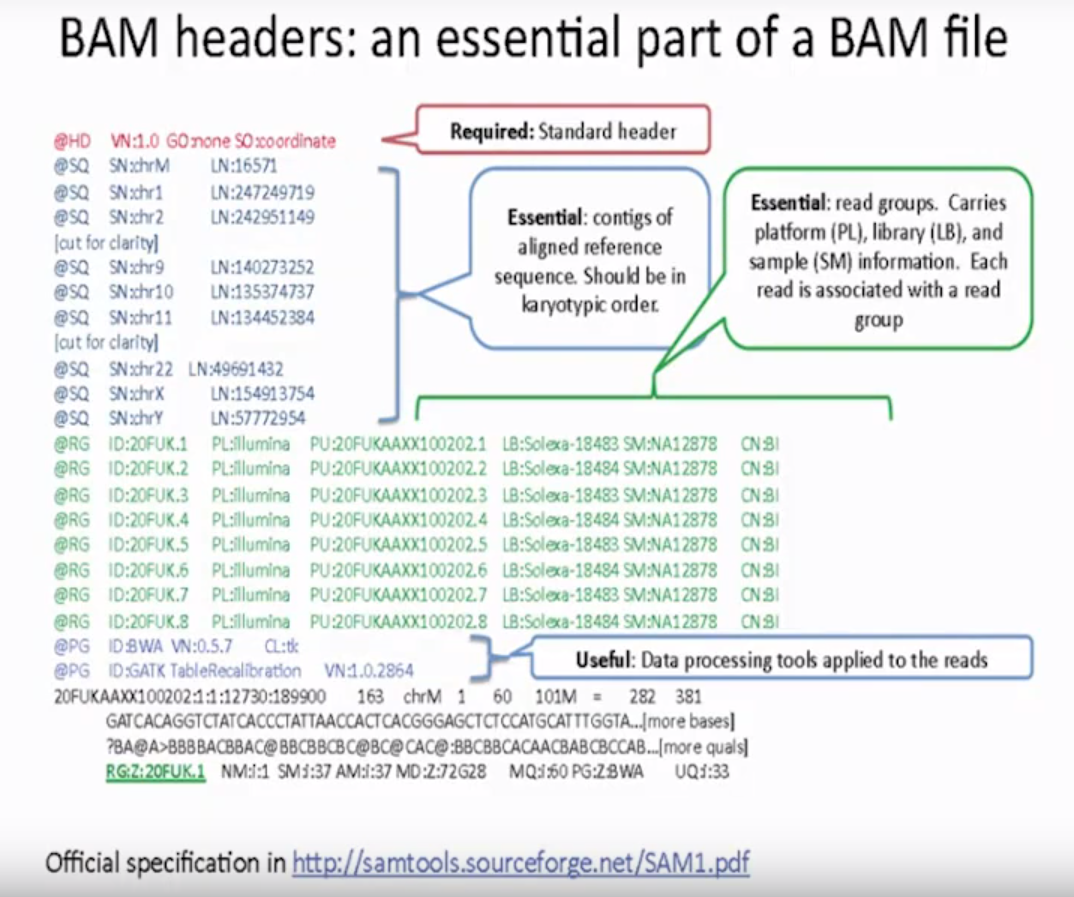
BAM headers: an essential part of a BAN file
================================================================================
BAM headers: an essential part of a BAN file
 ================================================================================
To deal with BAM file, use SAMtools
================================================================================
To deal with BAM file, use SAMtools
 ================================================================================
PySAM
================================================================================
PySAM
 ================================================================================
================================================================================
 ================================================================================
VCF files store variant information
================================================================================
VCF files store variant information
 ================================================================================
VCFtools
================================================================================
VCFtools
 ================================================================================
PyVCF
================================================================================
PyVCF
 ================================================================================
Generic Sequence Format (GSF) Version 3
================================================================================
Generic Sequence Format (GSF) Version 3

 ================================================================================
Parse GFF format file
================================================================================
Parse GFF format file
 ================================================================================
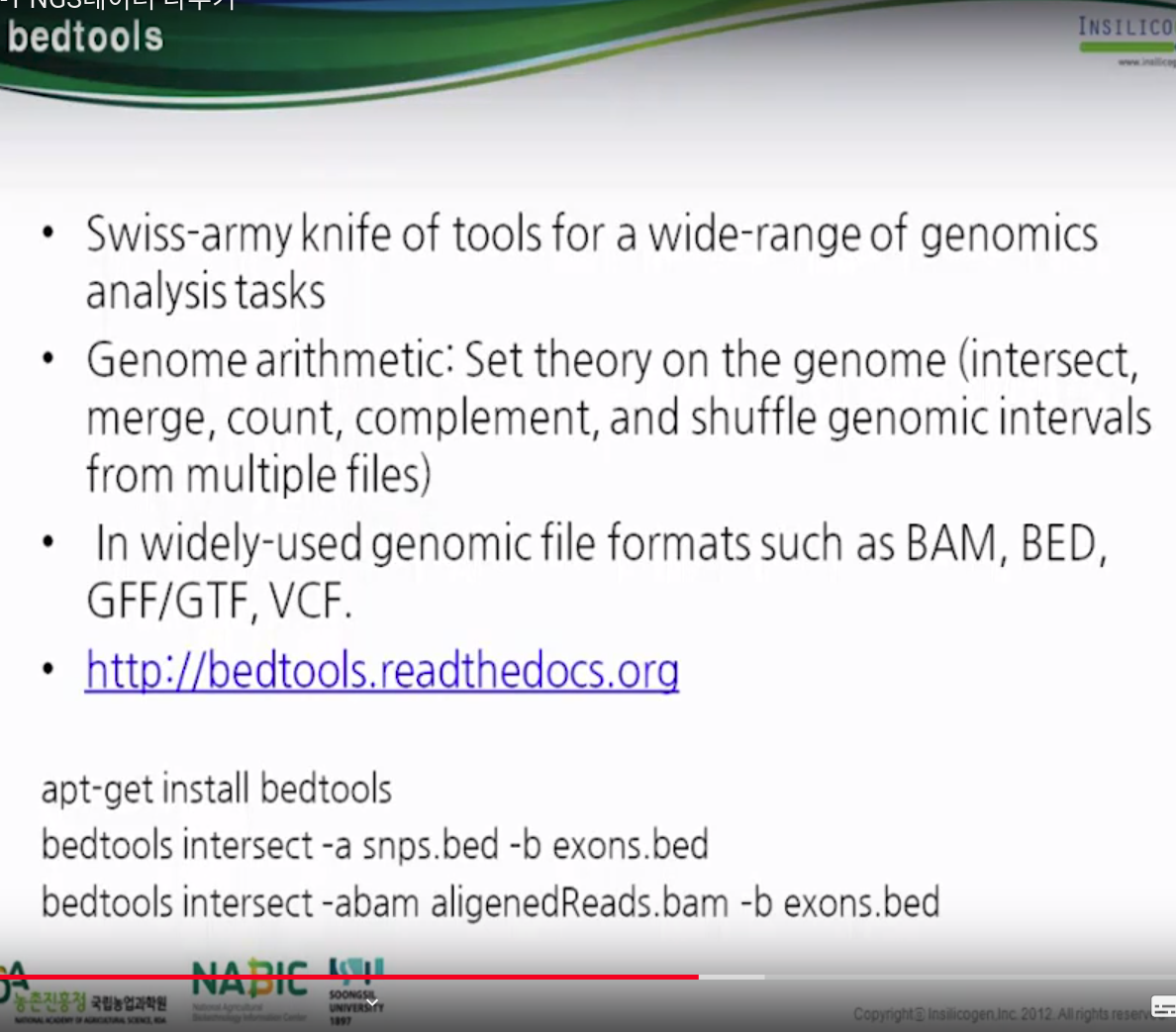
bedtools
================================================================================
bedtools
 ================================================================================
pybedtools
================================================================================
pybedtools
 ================================================================================
================================================================================

 ================================================================================
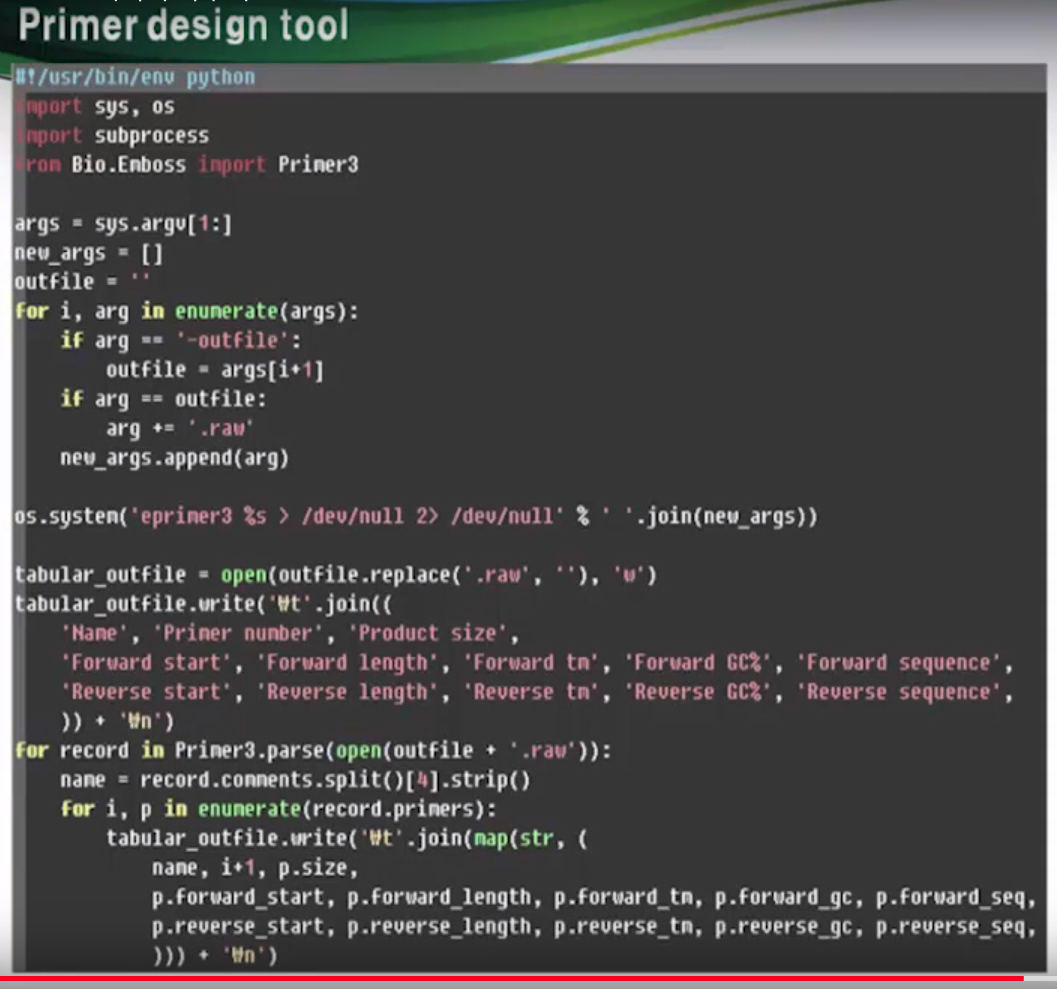
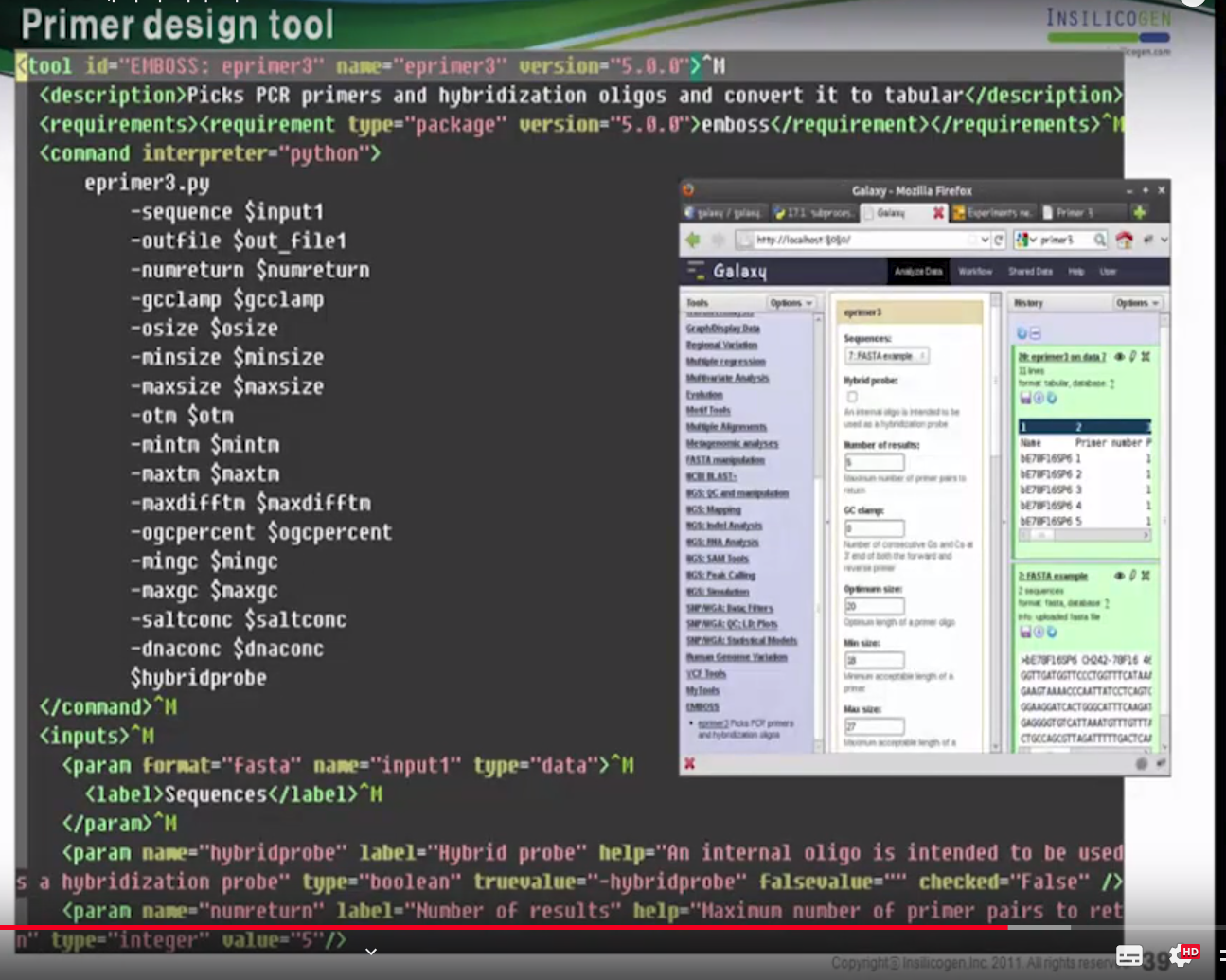
Primer design tool
================================================================================
Primer design tool